新型2,4-二取代四氢吡咯-3-羧酸衍生物的合成
2016-02-25章炜,徐亮
章 炜, 徐 亮
(四川大学 华西药学院 天然药物化学系,四川 成都 610041)
·快递论文·

通信联系人: 徐亮,副教授, Tel. 028-85503046, E-mail: liangxu@scu.edu.cn
新型2,4-二取代四氢吡咯-3-羧酸衍生物的合成
章炜, 徐亮*
(四川大学 华西药学院 天然药物化学系,四川 成都610041)
摘要:N-三甲硅甲基苯甲醛亚胺为非稳定型亚甲胺基叶立德前体,在磷酸催化下与取代噁唑烷酮烯烃经1,3-偶极环加成反应合成了10个具有cis-trans立体结构的新型2,3,4-三取代四氢吡咯-3-羧酸衍生物(3a~3j),收率65%~75%,其结构经1H NMR,13C NMR和HR-MS表征。3b的立体结构经X-单晶衍射确证。
关键词:N-三甲硅甲基苯甲醛亚胺; 非稳定型亚甲胺基叶立德; 1,3-偶极环加成; 四氢吡咯-3-羧酸; 合成
取代四氢吡咯类化合物具有高度的生物活性,如抗菌[1]、抗流感病毒[2]、酶抑制[3]和受体拮抗[4]等。其中,2,3,4-三取代四氢吡咯衍生物由于具有与内皮素受体A的显著选择性拮抗作用而受到学者的高度重视。如具有2,4-二取代四氢吡咯-3-羧酸结构的阿曲生坦及其异构体[5]。目前,阿曲生坦已进入治疗晚期前列腺癌的的III期临床研究阶段[6],对高血压代谢综合症、慢性肾病和非小细胞肺癌等疾病的临床研究也已取得一定成果[7-8]。
合成2,4-二芳基取代的四氢吡咯-3-羧酸衍生物的方法主要有:Michael加成[5,9],Mannich环化[10]和芳基锂与亚胺加成[11]等。这些方法均存在步骤长,选择性和收率偏低等缺点。1,3-偶极环加成反应是合成取代四氢吡咯最直接有效的方法。Tsuge O等[12]以N-硅甲基醛亚胺为原料,在醋酸和水作用下生成非稳定胺基叶立德,再与肉桂酸甲酯发生1,3-偶极环加成反应,合成了不易分离的三取代四氢吡咯顺反异构体混合物,收率约70%。本课题组用类似的方法进一步研究,发现该反应条件对于其它含富电子芳基取代的烯酯底物,收率也较差(约30%)。
为提高反应收率,本文以N-三甲硅甲基苯甲醛亚胺(1)为非稳定型亚甲胺基叶立德前体,在磷酸催化下与取代噁唑烷酮烯烃(2a~2j)经1,3-偶极环加成反应,合成了10个具有cis-trans立体结构的2,3,4-三取代四氢吡咯-3-羧酸衍生物(3a~3j, Scheme 1),收率65%~75%,其结构经1H NMR,13C NMR和HR-MS表征。3b的立体结构经X-单晶衍射确证。

Scheme 1
1实验部分
1.1 仪器与试剂
Varian Unity NOVA-400 MHz/54型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters Q-TOF-Premier型质谱仪。
1按文献[13]方法合成;硅胶,200~300目,青岛海洋化工厂;GF254硅胶板,烟台江友硅胶开发有限公司;其余所用试剂均为分析纯。
1.2 3a~3j的合成(以3a为例)
氩气保护下,在反应管中加入1 153 mg(0.8 mmol)和重蒸乙腈1 mL,搅拌使其溶解;加入苯取代噁唑烷酮烯烃(2a)87 mg(0.4 mmol)和磷酸2 μL,于室温反应至终点。加入乙酸乙酯2 mL,依次用饱和NaHCO3溶液和饱和食盐水洗涤,用无水Na2SO4干燥,蒸除溶剂后经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=1 ∶1]纯化得白色固体3a。
以2b~2j替代2a,用类似的方法合成白色固体3b~3f和无色油状液体3g~3j。
3a:1H NMRδ: 7.36~7.21(m, 10H), 4.91(d,J=10.0 Hz, 1H), 4.71(t,J=9.2 Hz, 10.0 Hz, 1H), 4.11~4.02(m, 2H), 3.71~3.58(m, 3H), 3.15(t,J=10.8 Hz, 1H), 3.01(m, 1H);13C NMRδ:172.1, 152.9, 141.1, 140.1, 128.6, 128.0, 127.8, 127.7, 127.6, 126.7, 66.2, 61.7, 56.9, 55.2, 48.0, 42.3; HR-MSm/z: Calcd for C20H20N2O3{[M+H]+}336.373 6, found 336.373 9。
3b:Rf=0.55(展开剂:A=1 ∶2,下同);1H NMRδ: 7.30(m, 8H), 6.99(t,J=8.8 Hz, 8.4 Hz, 2H), 4.90(d,J=10.2 Hz, 1H), 4.64(t,J=9.2 Hz, 9.6 Hz, 1H), 4.05(m, 2H), 3.66(m, 3H), 3.09(t,J=10.4 Hz, 10.8 Hz, 1H), 2.99(m, 1H);13C NMRδ: 171.9, 162.8, 160.4, 152.9, 140.1, 136.6, 129.2, 129.1, 128.0, 127.7, 127.6, 115.4, 115.2, 65.9, 61.7, 57.1, 55.0, 47.1, 42.3; HR-MSm/z: Calcd for C20H19N2O3F{[M+H]+}354.374 9, found 354.374 7。
3c:Rf=0.48;1H NMRδ: 7.39~7.29(m, 6H), 7.28~7.20(m, 2H), 7.16~7.10(m, 2H), 4.94(d,J=10.0 Hz, 1H), 4.83(t,J=8.8 Hz, 10.0 Hz, 1H), 4.36(m, 1H), 4.05(m, 1H), 3.64(m, 3H), 3.02(m, 1H), 2.49(s, 3H);13C NMRδ: 172.5, 152.9, 139.9, 139.3, 136.7, 130.4, 128.6, 128.0, 127.7, 127.6, 126.4, 126.4, 126.3, 66.3, 61.6, 56.7, 55.3, 43.5, 42.3, 19.9; HR-MSm/z: Calcd for C21H22N2O3{[M+H]+}350.411 0, found 350.411 4。
3d:Rf=0.45;1H NMRδ: 7.31(m, 4H), 6.85~6.79(m, 2H), 6.76(d,J=8.0 Hz, 1H), 5.93(s, 2H), 4.89(d,J=10.0 Hz, 1H), 4.63(t,J=9.6 Hz, 10.0 Hz, 1H), 4.10~4.00(m, 2H), 3.73~3.60(m, 3H), 3.11(d,J=10.8 Hz, 1H), 3.02(m, 1H);13C NMRδ: 172.2, 158.9, 158.3, 152.9, 132.9, 132.2, 128.7, 128.7, 113.9, 113.3, 65.4, 61.7, 56.9, 55.2, 55.1, 55.0, 47.2, 42.3; HR-MSm/z: Calcd for C21H20N2O5{[M+H]+}380.393 9, found 380.394 4。
3e:Rf=0.60;1H NMRδ: 8.41(d,J=8.4 Hz, 1H), 7.86(d,J=8.0 Hz, 1H), 7.76(d,J=8.0 Hz, 1H), 7.62(d,J=6.8 Hz, 1H), 7.58(m, 1H), 7.47(m, 2H), 7.42(d,J=6.8 Hz, 2H), 7.32(m,J=6.8 Hz, 7.6 Hz, 2H), 7.29(m, 1H), 5.09~4.92(m, 2H), 4.03(m, 1H), 3.86(dd,J=7.2 Hz, 7.2 Hz, 1H), 3.69(m, 1H), 3.58(m, 1H), 3.18(t,J=10.4 Hz, 10.4 Hz, 1H), 3.03(m, 1H);13C NMRδ: 172.6, 139.8, 137.5, 133.9, 132.1, 128.7, 128.1, 127.8, 127.6, 127.2, 126.1, 125.6, 123.7, 123.5, 66.5, 61.6, 56.2, 55.9, 42.9, 42.3; HR-MSm/z: Calcd for C24H22N2O3{[M+H]+}386.443 1, found 386.442 4。
3f:Rf=0.50;1H NMRδ: 7.29(m, 6H), 6.29(m, 1H), 6.13(d,J=2.8 Hz, 1H), 4.83(m, 2H), 4.17(m, 1H), 4.09(m, 1H), 3.71~3.64(m, 3H), 3.18(t,J=10.0 Hz, 10.4 Hz, 1H), 3.04(m, 1H);13C NMRδ: 172.2, 154.6, 152.8, 141.6, 139.3, 127.9, 127.7, 127.4, 110.0, 105.3, 66.0, 61.6, 53.9, 52.3, 42.3, 41.3; HR-MSm/z: Calcd for C18H18N2O4{[M+H]+}326.346 5, found 326.347 2。
3g:Rf=0.52;1H NMRδ: 7.32(d,J=4.0 Hz, 4H), 7.21(m, 2H), 7.17(t,J=3.2 Hz, 3.2 Hz, 1H), 6.95(d,J=3.2 Hz, 1H), 4.89(d,J=10.0 Hz, 1H), 4.67(t,J=9.2 Hz, 9.6 Hz, 1H), 4.37(m, 1H), 4.09(m, 1H), 3.07(m, 3H), 3.17(t,J=10.8 Hz, 10.4 Hz, 1H), 3.03(m, 1H);13C NMRδ: 171.7, 152.8, 144.4, 139.9, 128.0, 127.7, 127.6, 126.8, 124.3, 124.1, 123.5, 65.5, 61.7, 57.7, 55.2, 42.9, 42.3; HR-MSm/z: Calcd for C18H18N2O3S{[M+H]+}342.412 1, found 342.411 8。
3h:Rf=0.25;1H NMRδ: 7.30(m, 5H), 5.66(m, 2H), 4.55(m, 2H), 4.08(m, 1H), 3.67(m, 2H), 3.56(m, 1H), 3.03(m, 1H), 2.75(m, 2H), 2.20(m, 1H), 2.08(m, 3H), 1.79(m, 1H), 1.67(m, 1H), 1.28(m, 1H);13C NMRδ: 173.7, 139.0, 127.8, 127.4, 127.2, 127.2, 126.9, 126.8, 125.8, 125.7, 66.6, 61.4, 48.6, 48.3, 42.4, 37.6, 30.8, 30.2, 30.0, 27.1, 25.0; HR-MSm/z: Calcd for C20H24N2O3{[M+H]+}340.416 2, found 340.417 0。
3i:Rf=0.58;1H NMRδ: 7.25(m, 5H), 4.59(d,J=9.6 Hz, 1H), 4.30(t,J=7.2 Hz, 8.8 Hz, 1H), 4.10(m, 1H), 3.70~3.63(m, 2H), 3.51(m, 1H), 3.08(m, 1H), 2.82(m, 1H), 2.62(m, 1H), 1.50(m, 2H), 1.28(m, 6H), 0.87(m, 3H);13C NMRδ: 173.4, 162.4, 139.7, 127.8, 127.4, 127.2, 65.8, 61.5, 54.7, 53.2, 42.9, 42.4, 33.5, 32.3, 27.9, 22.4, 13.9; HR-MSm/z: Calcd for C19H26N2O3{[M+H]+}330.421 3, found 330.421 0。
3j:Rf=0.45;1H NMRδ: 7.28(m, 5H), 4.51(d,J=2.8 Hz, 2H), 4.08(m, 1H), 3.67(m, 2H), 3.51(m, 1H), 3.03(m, 1H), 2.71(m, 1H), 2.62(m, 1H), 1.65(m, 1H), 0.96(m, 6H);13C NMRδ: 173.1, 160.3, 139.1, 127.8, 127.4, 127.2, 66.8, 61.5, 54.7, 53.2, 42.9, 31.8, 27.6, 22.5, 14.0; HR-MSm/z: Calcd for C17H22N2O3{[M+H]+}302.368 2, found 302.368 0。
2结果与讨论
2.1 合成
以3i的合成为例,研究了多种质子酸催化剂(Cat)对3i总收率的影响,结果见表1。
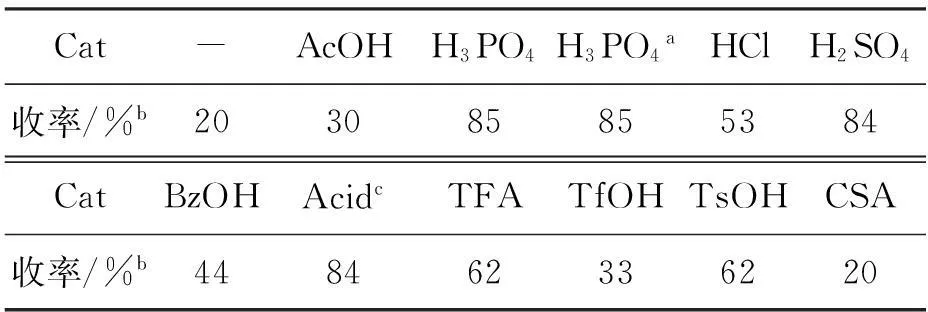
表 1 催化剂对3i收率的影响*
**Cat 10 mmol%, H2O 100 mol%,其余反应条件同1.2;a未加水;b总收率;cp-nitrobenzoic acid
由表1可见,磷酸的催化效果最好,总收率85%;是否加水,对磷酸的催化效果无影响。其可能原因在于:磷酸的多质子取代模式对烯烃底物的活化和偶极子的生成具有双重催化作用。
2.2 噁唑烷酮烯烃底物拓展
以H3PO4(10 mol%)为催化剂,乙腈为溶剂,其余反应条件同1.2,对底物(2b~2j)进行拓展,考察了该反应的底物普适性,结果见Scheme 1。由Scheme 1可见,芳基取代(2a~2c),芳杂环取代(2d, 2f, 2g)和稠环芳烃取代(2e)的噁唑烷酮烯烃,均能以较高收率(>70%)合成最终产物,脂肪烃基取代(2h~2j)的烯烃在此条件下合成的4-烷基取代四氢吡咯衍生物,收率也较高(>65%)。 2,3-cis与2,3-trans两种异构体的比例约为6 ∶1~8 ∶1。异构体可通过硅胶柱层析分离。
以3b为例,利用单晶X-射线单晶衍射对主要异构体的立体化学结构进行表征,结果见图1。
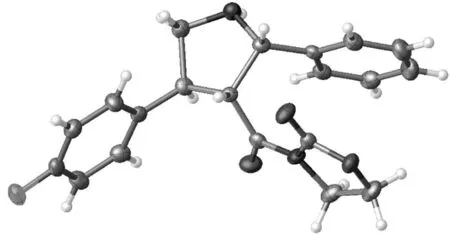
图 1 3b的晶体结构图
3结论
合成了10个具有cis-trans立体结构的新型2,3,4-三取代四氢吡咯-3-羧酸衍生物,收率较高(65%~75%)。该研究工作为寻找对ETA受体具有更高选择性与抑制活性的候选药物奠定了一定基础。
参考文献
[1]Hong C Y, Kim Y K, Chang J H,etal. Novel fluoroquinolone antibacterial agents containing oxime-substituted (aminomethyl) pyrrolidines:Synthesis and antibacterial activity of 7-[4-(aminomethyl)-3-(methoxyimino) pyrrolidin-1-yl]-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro[1,8]naphthyridine-3-carboxylic acid[J].J Med Chem,1997,40:3584-3593.
[2]Stylianakis I, Kolocouris A, Kolocouris N,etal. Spiro[pyrrolidine-2,2′-adamantanes]:Synthesis,anti-influenza virus activity and conformational properties[J].Bioorganic & Medicinal Chemistry Letters,2003,13:1699-1703.
[3]Pei Z H, Li X F, Longenecker K,etal. Discovery,structure,activity relationship and pharmacological evaluation of (5-substituted-pyrrolidinyl-2-carbonyl)-2-
cyanopyrrolidines as potent dipeptidyl peptidase IV inhibitors discovery,structure-activity relationship and pharmacological evaluationof (5-substituted-pyrrolidinyl-2-carbonyl)-2-cyanopyrrolidines as potent dipeptidyl peptidase IV inhibitors[J].J Med Chem,2006,49:3520-3535.
[4]Shih N Y, Aslanian R, Orlando S,etal. A novel pyrrolidine analog of histamine as a potent,highly selective histamine H3 receptor agonist[J].J Med Chem,1995,38:1593-1599.
[5]Winn M, Geldern T W, Opgenorth T J,etal. 2,4-Diarylpyrrolidine-3-carboxylic acids-potent ETA selective endothelin receptor Antagonists 1:Discovery of A-127722[J].J Med Chem,1996,39:1039-1048.
[6]Nelson J B, William L, Chin J L,etal. Phase 3,randomized,controlled trial of atrasentan in patients with nonmetastatic,hormone-refractory prostate cancer[J].Cancer,2008,113:2478-2487
[7]Raichlin E, Prasad A, Mathew V,etal. Efficacy and safety of atrasentan in patients with cardiovascular risk and early atherosclerosis[J].Hypertension,2008,52:522-528.
[8]Kohan D E, Pritchett Y, Molitch M,etal. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy[J].J Am Soc Nephrol,2011,22:763-772.
[9]Jae H S, Martin W, Thomas W G,etal. Pyrrolidine-3-carboxylic acids as endothelin antagonists 5:Highly selective,potent and orally active ETA antagonists[J].J Med Chem,2001,44:3978-3984.
[10]Wittenberger S J, McLaughlin M A. Preparation of endothelin antagonist ABT-627[J].Tetrahedron Letters,1999,40:7175-7178.
[11]Buchholz M, Reißig H U. Enantioselective synthesis of the pyrrolidine core of endothelin antagonist ABT-627(atrasentan) via 1,2-oxazines[J].Eur J Org Chem,2003,18:3524-3533.
[12]Tsuge O, Kanemasa S, Matsuda K. Synthetic versatility ofN-(silylmethyl) imines:Water-induced generation ofN-protonated azomethine ylides of nonstabilized type and fluoride-induced generation of 2-azaallyl anions[J].Bull Chem Soc Jpn,1986,59:2537-2545.
[13]Tsuge O, Kanemasa S, Matsuda K. A silyl-functionalized alkyl azide,trimethylsilyl-methyl azide:Synthesis and cycloaddition reaction to acetylenic dipolarophiles[J].Chemistry Letters,1983,12(7):1131-1134.
Synthesis of Novel 3,4-Disubstituted
Pyrrolidine-3-carboxylic Acid Derivatives
ZHANG Wei,XU Liang*
(Department of Natural Materia Medica, West China School of Pharmacy, Sichuan University, Chengdu 610041, China)
Abstract:Ten novel cis-trans-2,4-disubstituted pyrrolidine-3-carboxylic acid derivatives(3a~3j) in yield of 65%~75% were synthesized by 1,3-dipolar cycloaddition reaction of N-(trimethylsilylmethyl)benzaldehyde imine with α,β-unsaturated oxazolidinone amides, using phosphoric acid as catalyst. The structures were characterized by1H NMR,13C NMR and HR-MS. The stereochemical structure of 3b was confirmed by X-ray single crystal diffraction.
Keywords:N-(trimethylsilylmethyl)benzaldehyde imine; unstabilized azomethine ylide; 1,3-dipolar cycloaddition; pyrrolidine-3-carboxylic acid; synthesis
中图分类号:O623.731; O626.24
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.01.15099
作者简介:章炜(1990-),男,汉族,安徽芜湖人,硕士研究生,主要从事天然药物合成的研究。 E-mail: 948397896@qq.com
基金项目:国家自然科学基金资助项目(20902061)
收稿日期:2015-04-18;
修订日期:2015-11-19
