娃儿藤碱类似物DCB-3501的全合成及其细胞毒性
2016-02-25李松涛黄学石
李松涛, 刘 江, 黄学石
(1. 承德医学院 河北省中药研究与开发重点实验室,河北 承德 067000;
2. 中国医科大学 代谢病分子机制与药物研究所,辽宁 沈阳 110122)

娃儿藤碱类似物DCB-3501的全合成及其细胞毒性
李松涛1*, 刘江2, 黄学石2
(1. 承德医学院 河北省中药研究与开发重点实验室,河北 承德067000;
2. 中国医科大学 代谢病分子机制与药物研究所,辽宁 沈阳110122)
摘要:以3,4-二甲氧基苯甲醛与3,4-二甲氧基苯乙酸为起始原料,经Perkin缩合、自由基氧化偶联反应、Swern氧化、还原胺化及付克酰基化等7步反应全合成了娃儿藤碱类似物DCB-3501,其结构经1H NMR和ESI-MS确证。体外细胞毒性测试结果表明:DCB-3501对人结肠癌细胞HCT116、人胃癌细胞BGC-823、人肝癌细胞HepG-2、人宫颈癌细胞HeLa和人大细胞肺癌细胞H460的IC50分别为20.0 μmol·L-1, 50.9 μmol·L-1, 2.1 μmol·L-1, 65.8 μmol·L-1和30.8 μmol·L-1。
关键词:3,4-二甲氧基苯甲醛; 3,4-二甲氧基苯乙酸; 娃儿藤碱类似物; DCB-3501; Perkin缩合; Swern氧化; 付克酰基化; 全合成; 细胞毒性
娃儿藤碱(Tylophorine)及其类似物(Chart 1)是从罗藦科娃儿藤属和鹅绒藤属等植物中提取分离到的一类天然生物碱[1-2]。上世纪九十年代初,美国国家癌症研究所开展了一项针对60种人肿瘤细胞系的体外增殖抑制活性筛选实验,结果表明多种娃儿藤碱的天然产物具有潜在、广谱的
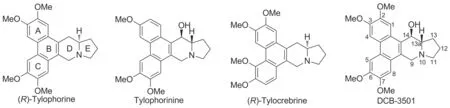
Chart 1
Scheme 1
抗肿瘤活性,GI50可达到纳摩级,有望成为新型抗肿瘤先导化合物。此后,该类生物碱及其类似物的化学合成、结构修饰及抗肿瘤活性日益成为研究热点[2-5]。
DCB-3501是Tylophorine C-14位氢被(R)-OH取代的类似物(Chart 1),由Rapoport H等[6]首次合成。该方法的关键步骤是利用谷氨酸二异丙酯与2,3,6,7-四甲氧基-9-菲醛反应引入N-谷氨酸二异丙酯侧链,该侧链经环化后得到E环,再进一步经水解与付克酰基化反应构建D环,最后D, E环上的羰基分别经过还原得到DCB-3501。 Cheng等[7-8]研究结果表明DCB-3501对人肝癌细胞HepG-2、鼻咽癌细胞KB均具有体外增殖抑制活性,EC50分别为(110±45)nmol·L-1和(106±84)nmol·L-1。 Wang等[9]发现DCB-3501对人肺癌细胞A549、人白血病细胞HL60也表现出一定的增殖抑制活性。
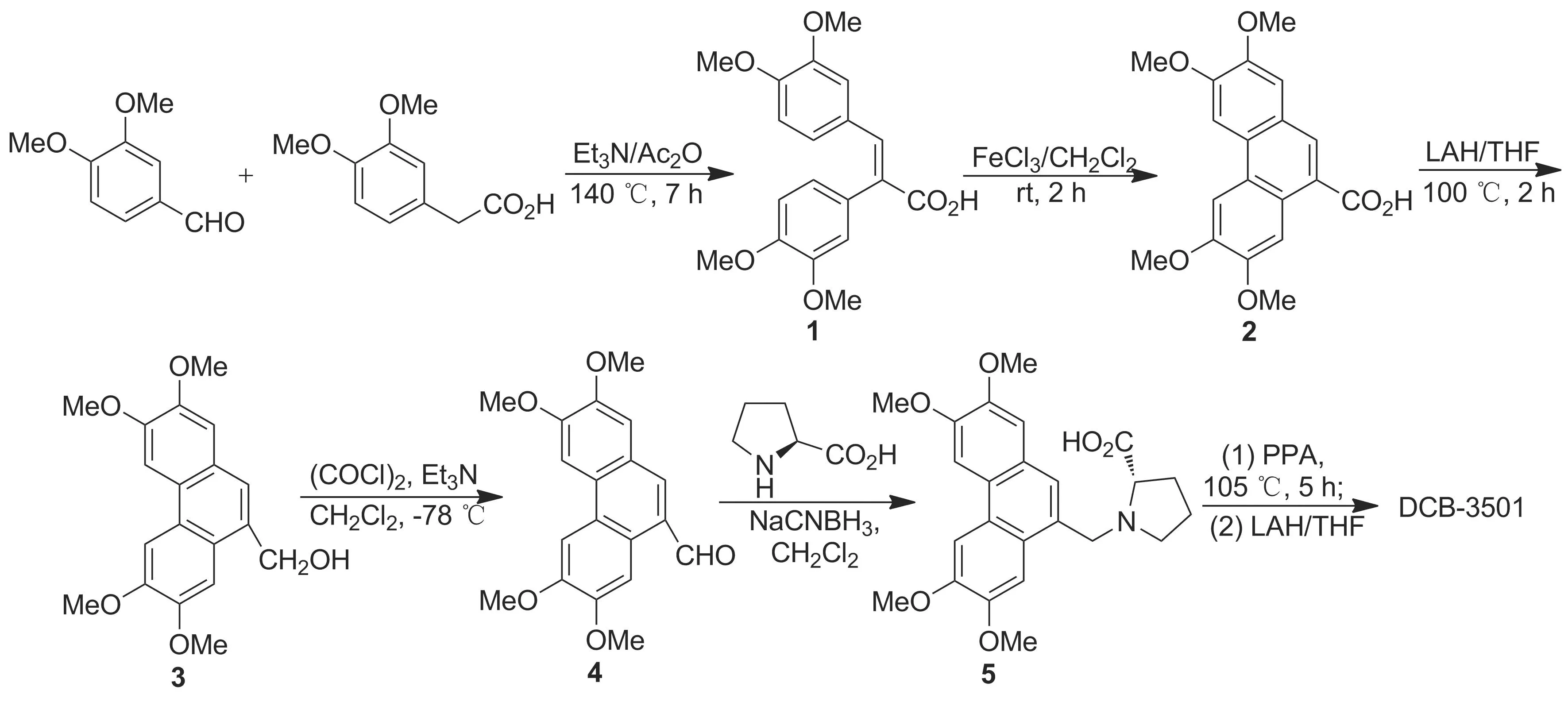
本文以3,4-二甲氧基苯甲醛与3,4-二甲氧基苯乙酸为起始原料,经Perkin缩合、自由基氧化偶联反应、Swern氧化、还原胺化及付克酰基化等7步反应实现了DCB-3501的全合成(Scheme 1),其结构经1H NMR和ESI-MS确证。并考察了其对人结肠癌细胞HCT116等五种肿瘤细胞株的体外细胞毒性。
1实验部分
1.1 试剂与仪器
Bruker ARX-300型或Bruker AV-600型核磁共振仪(CDCl3为溶剂,TMS为内标);Finnigan LCQ型质谱仪。
1.2 合成
(1)α-(3,4-二甲氧基苯基)-3,4-二甲氧基肉桂酸(1)的合成
在反应瓶中依次加入3,4-二甲氧基苯甲醛1.7 g(10 mmol), 3,4-二甲氧基苯乙酸1.9 g(10 mmol),三乙胺3 mL和醋酸酐20 mL,搅拌下回流(140 ℃)反应7 h。冷却至室温,加入水100 mL,回流30 min;冷却至室温,用乙酸乙酯(3×300 mL)萃取,合并萃取液,用饱和食盐水洗涤至水层呈中性,有机相用无水硫酸钠干燥,减压浓缩,用无水乙醇重结晶得深黄色粉末固体1 3.1 g,收率90%;1H NMRδ: 3.47(s, 3H, OCH3), 3.81(s, 3H, OCH3), 3.84(s, 3H, OCH3), 3.89(s, 3H, OCH3), 6.55(d,J=1.8 Hz, 1H, ArH), 6.70(d,J=1.8 Hz, 1H, ArH), 6.72(d,J=8.5 Hz, 1H, ArH), 6.84(dd,J=8.2 Hz, 1.8 Hz, 1H, ArH), 6.85(dd,J=8.5 Hz, 1.8 Hz, 1H, ArH), 6.92(d,J=8.2 Hz, 1H, ArH), 7.86(s, 1H, C=CH), 12.44(s, 1H, CO2H); ESI-MSm/z: 343{[M-H]-}。
(2) 2,3,6,7-四甲氧基-9-菲酸(2)的合成
在反应瓶中依次加入1 3.0 g(8.6 mmol)和无水二氯甲烷75 mL,搅拌使其溶解;氮气保护,加入无水三氯化铁5.8 g(36.7 mmol),于室温反应8 h。加甲醇20 mL,搅拌10 min,减压浓缩,加入甲醇20 mL,过滤,滤饼用甲醇(3×10 mL)洗涤,干燥得黄绿色固体2 2.1 g,收率70%;1H NMRδ: 3.90(s, 3H, OCH3), 3.91(s, 3H, OCH3), 4.06(s, 3H, OCH3), 4.11(s, 3H, OCH3), 7.55(s, 1H, ArH), 8.00(s, 1H, ArH), 8.03(s, 1H, ArH), 8.42(s, 1H, ArH), 8.55(s, 1H, ArH), 12.88(br s, 1H, CO2H)。
(3) 2,3,6,7-四甲氧基-9-菲醇(3)
冰盐浴冷却下,在2 1.78 g(5.2 mmol)的无水THF(100 mL)溶液中加入氢化锂铝1.19 g(31.2 mmol),反应30 min;回流(100 ℃)反应2 h。冷却至室温,加入冰块和饱和碳酸氢钠溶液(100 mL),搅拌10 min,分液,水相用THF(2×50 mL)萃取,合并有机相和萃取液,依次用饱和食盐水(200 mL)洗涤,无水硫酸钠干燥,减压浓缩后经硅胶柱层析[洗脱剂:A=V(二氯甲烷) ∶V(甲醇)=20 ∶1]纯化得白色固体3 1.19 g,收率70%;1H NMRδ: 3.97(s, 3H, OCH3), 4.03(s, 3H, OCH3), 4.08(s, 3H, OCH3), 4.10(s, 3H, OCH3), 5.05(s, 2H, CH2), 7.10(s, 1H, ArH), 7.47(s, 1H, ArH), 7.48(s, 1H, ArH), 7.68(s, 1H, ArH), 7.74(s, 1H, ArH)。
(4) 2,3,6,7-四甲氧基-9-菲醛(4)的合成
将干式变的冷却方式由自然风冷(AN)改为强迫空气冷却(AF),即通过风机将空气强迫吹进变压器散热气道,增强气道空气对流。
在反应瓶中加入无水二氯甲烷20 mL,丙酮-干冰浴冷却,保持温度在-78 ℃以下搅拌10 min,加入草酰氯1.6 mL(18.72 mmol),反应20 min;缓慢滴加二甲基亚砜3 mL(42 mmol)的无水二氯甲烷(20 mL)溶液,滴毕(20 min,保持温度在-78 ℃),反应30 min;缓慢滴加3 1.0 g(3.04 mmol)的二氯甲烷(20 mL)/二甲基亚砜(1.5 mL)溶液,滴毕(30 min),反应30 min;滴入三乙胺11 mL(78 mmol),滴毕(20 min),继续低温反应30 min。升至室温,加入1 mol·L-1盐酸100 mL,剧烈搅拌10 min,调节水层pH呈酸性,分出有机层,水层用二氯甲烷(2×50 mL)萃取,合并有机层和萃取液,用无水硫酸钠干燥,蒸干,用甲醇重结晶得黄色固体4 0.70 g,收率70%;1H NMRδ: 4.06(s, 3H, OCH3), 4.10(s, 3H, OCH3), 4.12(s, 3H, OCH3), 4.15(s, 3H, OCH3), 7.28(s, 1H, ArH), 7.72(s, 1H, ArH), 7.74(s, 1H, ArH), 8.01(s, 1H, ArH), 8.94(s, 1H, CHO)。
(5)N-(2,3,6,7-四甲氧基菲基-9-亚甲基)-L-脯氨酸(5)的合成
将L-脯氨酸431 mg(3.75 mmol)溶于甲醇(10 mL)中,加入甲醇/甲醇钠溶液,调至pH≈7,搅拌下缓慢滴加4 500 mg(1.5 mmol)的二氯甲烷(30 mL)溶液,滴毕,反应30 min,加入NaBH3CN 300 mg(4.5 mmol),于室温反应20 h。减压蒸干,经硅胶柱层析(梯度洗脱剂:A=50 ∶1~1 ∶1)纯化得白色固体5 0.61 g,收率60%;1H NMRδ: 1.91(m, 2H, CH2), 2.12(m, 2H, CH2), 2.99(d,J=12.0 Hz, 1H, ArCH2N), 3.39(m, 1H, CH2N), 3.47(m, 1H, CH2N), 3.90(s, 3H, OCH3), 4.02(s, 3H, OCH3), 4.07(s, 3H, OCH3), 4.17(s, 3H, OCH3), 4.52(m, 1H, CHN), 4.97(d,J=12.0 Hz, 1H, ArCH2N), 7.12(s, 1H, ArH), 7.53(s, 1H , ArH), 7.59(s, 1H, ArH), 7.64(s, 1H, ArH), 7.65(s, 1H, ArH); ESI-MSm/z: 426{[M+H]+}。
(6) DCB-3501的合成
在反应瓶中加入5 220 mg(0.5 mmol),氮气保护下于室温加入多聚磷酸(PPA, 5 mL),于105 ℃反应5 h。冷却至室温,加入1 mol·L-1KOH溶液50 mL,调至pH 10,用二氯甲烷萃取,萃取液用等量饱和食盐水洗涤,无水硫酸钠干燥,蒸干溶剂得黄褐色固体,用无水THF 10 mL溶解,搅拌下于室温加入氢化锂铝50 mg(1.3 mmol),反应1 h。加入二氯甲烷(30 mL)和饱和碳酸钠溶液(30 mL),剧烈搅拌10 min,静置,分离有机层,水相用二氯甲烷(2×50 mL)萃取,合并萃取液和有机相,用无水硫酸钠干燥,蒸干后经硅胶柱层析(洗脱剂:A=50 ∶1)纯化得灰白色固体DCB-3501 40 mg,收率20%;1H NMRδ: 0.87(m, 1H, 12-H), 1.89(m, 1H, 13-H), 2.05(m, 1H, 12-H), 2.26(m, 1H, 13a-H), 2.34(m, 1H, 11-H), 2.40(m, 1H, 13-H), 3.00 (d,J=11.1 Hz, 1H, 9-H), 3.36(m, 1H, 11-H), 3.37 (d,J=11.1 Hz, 1H, 9-H), 3.80(s, 3H, OCH3), 4.08(s, 3H, OCH3), 4.10(s, 3H, OCH3), 4.14(s, 3H, OCH3), 4.86(br s, 1H, 14-H), 5.28(s, 1H, 14-OH), 6.08(s, 1H, ArH), 7.39(s, 1H, ArH), 7.59(s, 1H, ArH), 7.85(s,1H, ArH); ESI-MSm/z: 410{[M+H]+}。
1.3 体外细胞毒性测定
采用MTT法[10]测定DCB-3501对人结肠癌细胞HCT116、人胃癌细胞BGC-823、人肝癌细胞HepG-2、人宫颈癌细胞HeLa和人大细胞肺癌细胞H460的体外细胞毒性,阿霉素为对照药物。用DMSO溶解DCB-3501,用培养基稀释制得六个不同浓度[(1.0, 0.33, 0.10, 0.033, 0.010和0.0033) mmol·L-1]溶液,分别取六种浓度的溶液10 μL加至含有90 μL培养基(约5 000个细胞)的板孔中,于37 ℃孵育72 h,每孔加入10 μL 的MTT溶液 (5 mg·mL-1),于37 ℃孵育4 h后,弃去上清液,每孔加入150 μL DMSO,在570 nm处,用酶标仪测量每个孔的OD值,根据公式计算抑制率和相应的IC50。
2结果与讨论
2.1 合成
在将二苯乙烯类衍生物合成菲环(即由1合成2)的过程中,我们首先使用三氟醋酸铊介导二苯乙烯类衍生物1关环[11-12],通过实验条件的探索优化, 2的收率85%左右,但会形成菲环的多聚体;文献[13]报道m-CPBA/TFA系统能够有效介导二苯乙烯类衍生物的氧化偶联反应,我们尝试了该方法,没有得到目标关环产物。Wang等[14]采用过量的无水三氯化铁介导二苯乙烯类衍生物合成菲环,反应条件温和,后处理简便,本文选择该方法合成2。
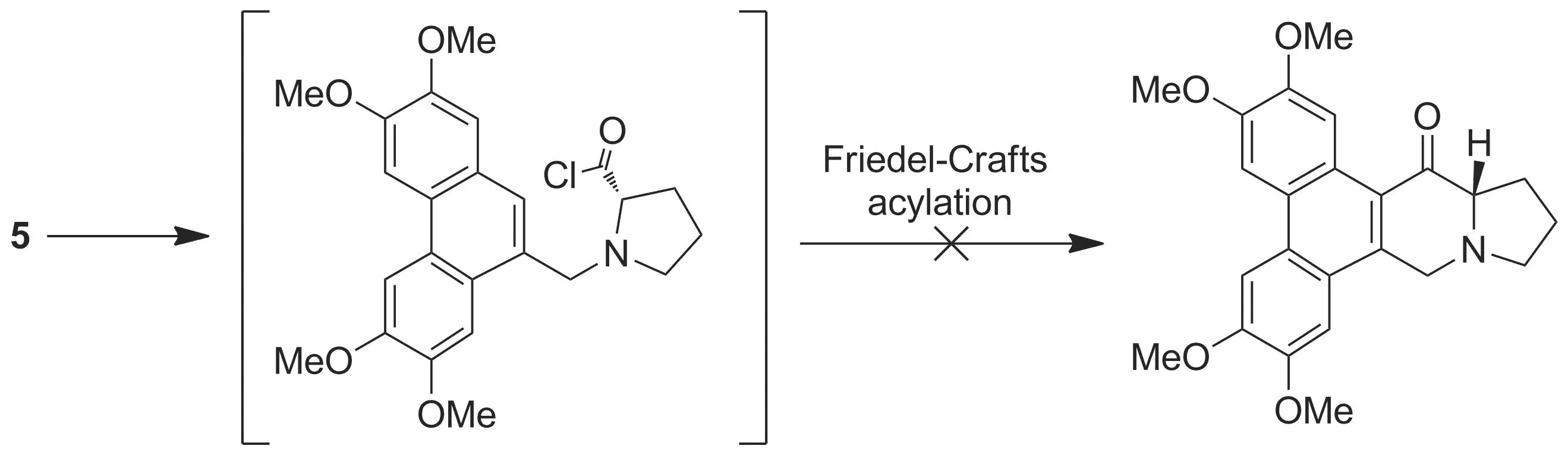
在尝试将5应用付克酰基化反应合成吲哚里西丁时,本文分别使用路易斯酸无水AlCl3和无水FeCl3作为催化剂,但是均未得到目标吲哚环关环产物(Scheme 2),与Rapoport H等[6]的实验结果一致。
多聚磷酸(PPA)是一种常用的酰化试剂,Chauncy B等[15]将N-菲-9-亚甲基-L-脯氨酸在PPA中加热,L-脯氨酸的羧基与菲环发生付克酰基化反应得到酮式中间体。参考该方法,我们通过摸索反应条件使5在PPA的作用下反应合成关键的酮式中间体。
Rapoport H等利用谷氨酸二异丙酯在菲环9-位引入N-谷氨酸二异丙酯侧链,该侧链经环化后构建DCB-3501的E环,再经水解与付克酰基化反应构建D环,D、E环上的羰基分别经过还原得到DCB-3501。本实验通过2,3,6,7-四甲氧基-9-菲醛与L-脯氨酸反应直接在菲环9-位引入E环,再经付克酰基化反应和一步羰基还原反应得到DCB-3501,简化了反应步骤。
Scheme 2

CompIC50/μmol·L-1HCT116BGC-823HepG-2HeLaH460DCB-350120.050.92.165.830.8 Adriamycinhydrochloride1.41.50.51.01.0
2.2 体外细胞毒性
DCB-3501对五种肿瘤细胞的体外细胞毒性结果见表1。由表1可见,DCB-3501对HepG-2细胞的细胞毒性最强(IC50=2.1 μmol·L-1),对HCT116等其他四种细胞具有一定的增殖抑制活性(IC50=20.0~65.8 μmol·L-1),表明其对不同来源瘤株的敏感性存在差异,但对相应细胞的细胞毒性均弱于对照药物阿霉素(IC50=0.5~1.4 μmol·L-1)。
3结论
以取代苯甲醛与苯乙酸为原料,经7步反应全合成了娃儿藤碱类似物DCB-3501。体外细胞毒性实验结果表明:DCB-3501对不同来源肿瘤细胞的敏感性存在差异,对HepG-2细胞的细胞毒性最强(IC50=2.1 μmol·L-1)。
该研究工作为娃儿藤碱及其类似物的抗肿瘤活性研究提供参考。
参考文献
[1]Gellert E. The indolizidine alkaloids[J].J Nat Prod,1982,45:50-73.
[2]Li Z G, Jin Z, Huang R Q. Isolation,total synthesis and biologicalactivity of phenanthroindolizidine and phenanthroquinolizidine alkaloids[J].Synthesis,2001,16:2365-2378.
[3]Chemler S E. Phenanthroindolizidines and phenanthroquinolizidines:Promising alkaloids for anti-cancer therapy[J].Curr Bioact Compd,2009,5(1):2-19.
[4]张成刚,谭显东. 菲并吲哚里西定类生物碱的研究进展[J].四川师范大学学报(自然科学版),2005,28(3):366-370.
[5]王远兴,方志杰,高军峰,等. 由藜芦醛合成娃儿藤碱[J].应用化学,2007,24(2):215-219.
[6]Buckley T F, Rapoport H.α-Amino acids as chiral educts for asymmetric products.Chirally specific synthesis of tylophorine and cryptopleurine[J].J Org Chem,1983,48:4222-4232.
[7]Gao W, Lam W, Zhong S,etal. Novel mode of action of tylophorine analogs as antitumor compounds[J].Cancer Res,2004,64:678-688.
[8]Gao W, Busson S, Grill S P,etal. Structure-activity studies of phenanthroindolizidine alkaloids as potential antitumor agents[J].Bioorg Med Chem Lett,2007,17:4338-4342.
[9]Wang Z W, Wu M, Wang Y,etal. Synthesis and SAR studies of phenanthroindolizidine and phenanthroquinolizidine alkaloids as potent anti-tumor agents[J].European Journal of Medicinal Chemistry,2012,51:250-258.
[10]Li S T, Han L, Sun L,etal. Synthesis and antitumor activities of phenanthrene-based alkaloids[J].Molecules,2009,14:5042-5053.
[11]Cragg J E, Herbert R B. Synthesis of the alkaloids,3′,4′-dimethoxy-2-(2-piperidyl) acetophenone,julandine,and cryptopleurine[J].J Chem Soc Perkin Trans 1,1982,(10):2487-2490.
[12]Iwasa K, Wiggauchi M, Takao N. The preparation of the biosynthetic precursor 3,7-dihydroxy-2,6-dimethoxy-phenanthroindolizidine[J].J Nat Prod,1988,51:172-175.
[13]Wang K L, Hu Y N, Wu M,etal.m-CPBA/TFA:An efficient nonmetallic reagent for oxidative coupling of 1,2-diarylethylenes[J].Tetrahedron,2010,66:9135-9140.
[14]Wang K L, Lü M Y, Wang Q M,etal. Iron(III) chloride-based mild synthesis of phenanthrene and its application to total synthesis of phenanthroindolizidine alkaloids[J].Tetrahedron,2008,64:7504-7510.
[15]Chauncy B, Gellert E, Trivedi K N. A new synthesis of phenanthroindolizidine[J].Aust J Chem,1969,22:427-429.
·快递论文·
Total Synthesis and Cytotoxicity of Tylophorine Analogue DCB-3501
LI Song-tao1*,LIU Jiang2,HUANG Xue-shi2
(1.Hebei Key Laboratory of Study and Exploitation of Traditional Chinese Medicine, Chengde Medical
University, Chengde 067000, China; 2. Institute of Metabolic Disease Research and Drug Development,
China Medical University, Shenyang 110122, China)
Abstract:The tylophorine analogue DCB-3501 was totally synthesized by a seven-step reaction including Perkin condensation, free radical oxidative coupling, Swern oxidation, reductive amination and Fridel-Crafts acylation, using 3,4-dimethoxy-benzaldehyde and 3,4-dimethoxy-phenylacetic acid as the raw materials. The structure was confirmed by1H NMR and ESI-MS. The in vitro cytotoxicity results showed that the IC50of DCB-3501 against human colon cancer cell HCT116, human gastric cancer cell BGC-823, human hepatic cancer cell HepG-2, human cervical cancer cell HeLa and human large-cell lung cancer cell H460 were 20.0 μmol·L-1, 50.9 μmol·L-1, 2.1 μmol·L-1, 65.8 μmol·L-1and 30.8 μmol·L-1, respectively.
Keywords:3,4-dimethoxy-benzaldehyde; 3,4-dimethoxy-phenylacetic acid; tylophorine analogue; DCB-3501; Perkin condensation; Swern oxidation; Fridel-Crafts acylation; total synthesis; cytotoxicity
中图分类号:O621.3; O626
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.01.15279
作者简介:李松涛(1983-),汉族,辽宁朝阳人,博士,助理研究员,主要从事有机合成化学研究。 Tel. 0314-2290640, E-mail: songtao-li@hotmail.com
基金项目:国家自然科学基金资助项目(30600783); 承德医学院博士基金资助项目(201303)
收稿日期:2015-07-27
