QuEChERS-HPLC-MS/MS法检测液态乳中9种β2-受体激动剂残留量
2016-02-20
(西北农林科技大学食品科学与工程学院,农业部食品质量监督检验测试中心(杨凌),陕西杨凌712100)
QuEChERS-HPLC-MS/MS法检测液态乳中9种β2-受体激动剂残留量
李婧妍,郭春锋,崔立辉,刘拉平
(西北农林科技大学食品科学与工程学院,农业部食品质量监督检验测试中心(杨凌),陕西杨凌712100)
建立了QuEChERS-液相色谱串联质谱法同时检测液态乳中9种β2-受体激动剂药物残留量方法。样品以乙腈、EDTA-Mcllvaine缓冲液和三氯乙酸为提取试剂、NaCl为吸水剂,PSA为吸附剂进行净化。用Shim-pack XR-ODS(3.0mmi.d× 75mm)色谱柱分离,以乙腈溶液和0.1%甲酸水溶液为流动相进行梯度洗脱,电喷雾正离子模式(ESI+)电离,多反应监测(MRM)模式检测。结果表明,样品经QuEChERS处理后,9种β2-受体激动剂在1.0~200 μg/kg范围内具有良好的线性关系,相关系数(r2)大于0.991,检出限(LOD)范围为0.03~0.13 μg/kg,定量限(LOQ)范围在0.1~0.4 μg/kg之间。回收率介于78.2%~96.0%之间,相对标准偏(RSD)均低于13.7%之间。
液态乳;β2-受体激动剂药物残留量;QuEChERS;高效液相色谱-串联质谱
0 引言
β2-受体激动剂是一类乳制品中经常能够检出的兽药之一,具有促进动物体内脂肪分解和提高瘦肉率等功效[1]。目前,检测-受体激动剂的方法有GC-MS[2]、HPLC-MS/MS[3]、ELISA[4]和HPLC[5-7]。我国和国际上常用的β2-受体激动剂确证方法为HPLC-MS/MS法,但该方法前处理环节存在有机溶剂消耗量大、操作复杂等缺陷。因此,需建立高通量、高灵敏度、简单快速的针对液态乳中β2-受体激动剂残留的专项方法。
QuEChERS提取方法在兽药残留检测方面得到广泛引用[8]。利用该方法,研究人员已有效提取液态乳中固醇类激素[9]和β2-内酰胺类[10]残留,但关于提取β2-受体激动剂的方法还未见报道。因此本研究用QuEChERS方法进行前处理,HPLC-MS/MS法检测液态乳中的β2-受体激动剂类兽药残留量。
1 实验
1.1仪器
高效液相色谱仪(岛津LC-2010A)-串联质谱仪(AB 4000 QTRAR),配有电喷雾(ESI)离子源;涡旋混合器(KQ-250DE);高速冷冻离心机;十万分之一分析天平;氮吹水浴浓缩仪(DCY-III)。
1.2标准品和试剂
克伦特罗、沙丁胺醇、莱克多巴胺、特布他林标准品(纯度≥99%);西马特罗、非诺特罗、氯丙那林、妥布特罗、喷布特罗标准品(纯度纯度≥99%)。C18、PSA、NH2和中性氧化铝吸附剂购自天津博纳艾杰尔公司。乙腈、甲醇为色谱纯;其他试验用试剂均为分析纯;试验所用液态乳。实验室用水为去离子水。
1.3色谱条件
色谱柱为 Shim-pack XR-ODS(3.0mmi.d× 75mm);流动相A为0.1%(体积分数)甲酸水,流动相B为0.1%甲酸乙腈。梯度洗过程序:0 ~1.5 min,流动相B为90%(体积分数);1.5 ~9 min,流动相B由90%降至5%;9 ~11 min,流动相B由5%升至90%;11.1 ~15 min,B为90%。流速为0.3 mL/min;进样量10 μL。ESI源正离子扫描;动态多反应监测。
1.4标准溶液配制
精确称取各种标准10.0 mg,分别置于100 mL容量瓶中,用甲醇稀释至刻度,即为质量浓度为0.1 mg/mL的标准储备液。吸取上述溶液各1 mL加入到100 mL容量瓶中,用甲醇稀释至刻度,即质量浓度为1.0 μg/mL的混合标准物质中间贮备液。吸取中间贮备液1 mL,用甲醇定容至100 mL,即为9种β2-受体激动剂质量浓度为10 ng/mL的标准溶液。
1.5样品前处理方法
称取样品2 g液态乳样品置于50 mL离心管中,加入2 mL浓度为0.1 mol/L的EDTA-Mcllvaine缓冲液、8 mL乙腈溶液和1 mL三氯乙酸,涡旋混合2 min,以10 000 r/min离心5 min。将上清液移入另一50 mL离心管中,加入6 g的Na2SO4,涡旋混合2 min,以10 000 r/min离心5 min。吸取2 mL提取液,加入50 mg PSA吸附剂,涡旋混合2 min,4 000 r/min离心5 min。移取上清液至10 mL离心管中,氮气吹干,加入1 mL体积比为10∶90的甲醇-0.1%甲酸水溶液溶解残渣,用0.45 μm再生纤维素膜过滤至样品瓶中。
1.6基质标准曲线制备
以空白样品提取液作为标准溶液的稀释液,配置质量分数分别为1.0,2.5,5.0,10,20,50,100,200,300 μg/kg的基质标准曲线。以各组分进样浓度为横坐标,峰面积为纵坐标绘制线性关系图,制作回归方程。以3倍和10倍信噪比(S/N)考察9种β2-受体激动剂的检出限(LOD)和定量限(LOQ)。
1.7方法回收率及检出限
称取阴性待测样品2 g,加入适量的混合标准溶液,配置成加标量分别为50,100和200 ng/g三个梯度的加标样品,依据优化的样品前处理方法和色谱条件进行测定,每个浓度平行测定6份样品,计算日内和日间回收率和相对标准偏差(RSD)。
2 结果与分析
2.1仪器条件优化
2.1.1 色谱条件优化
在分析本研究涉及的9种β2-受体激动剂时,研究人员多采用C18色谱柱进行分析[11-12]。因此本试验选用性能较好的Shim-pack XR-ODS(3.0 mmi.d×75 mm)色谱柱。流动相的使用对目标分析物的影响较大,相关研究中所用流动相多为水相和有机相的混合物,其中有机相为甲醇或乙腈,水相则为低浓度的甲酸水溶液,并执行梯度洗脱程序。本研究在确定色谱柱后对比了水-甲醇、水-乙腈、0.1%甲酸水-甲醇、0.1%甲酸水-乙腈、水-0.1%甲酸甲醇、水-0.1%甲酸乙腈、0.1%甲酸水-0.1%甲酸甲醇和0.1%甲酸水-0.1%甲酸乙腈组成的流动相的分析效果。结果表明,0.1%甲酸水-0.1%甲酸乙腈作为流动相时保留时间和分离效果相对较好,通过改变不同时间段水相和有机相比例,反复试验后确定最佳梯度洗脱条件为:0 ~1.5 min,流动相B的体积分数为90%;1.5 ~9 min,B由90%降至5%;9 ~11 min,B由5%升至90%;11.1 ~15 min,B为90%。
2.1.2 质谱条件优化
ESI+模式下分别对1 mg/L 9种β2-受体激动剂的标准溶液进行一级质谱扫描分析,得到每种成分的母离子,对其进行全扫描确定子离子。以MRM模式进行监测,优化每种β2-受体激动剂的母离子和子离子所需最佳碰撞能量(DP)和最佳锥孔电压(CE),得到优化的质谱参数如表1所示。
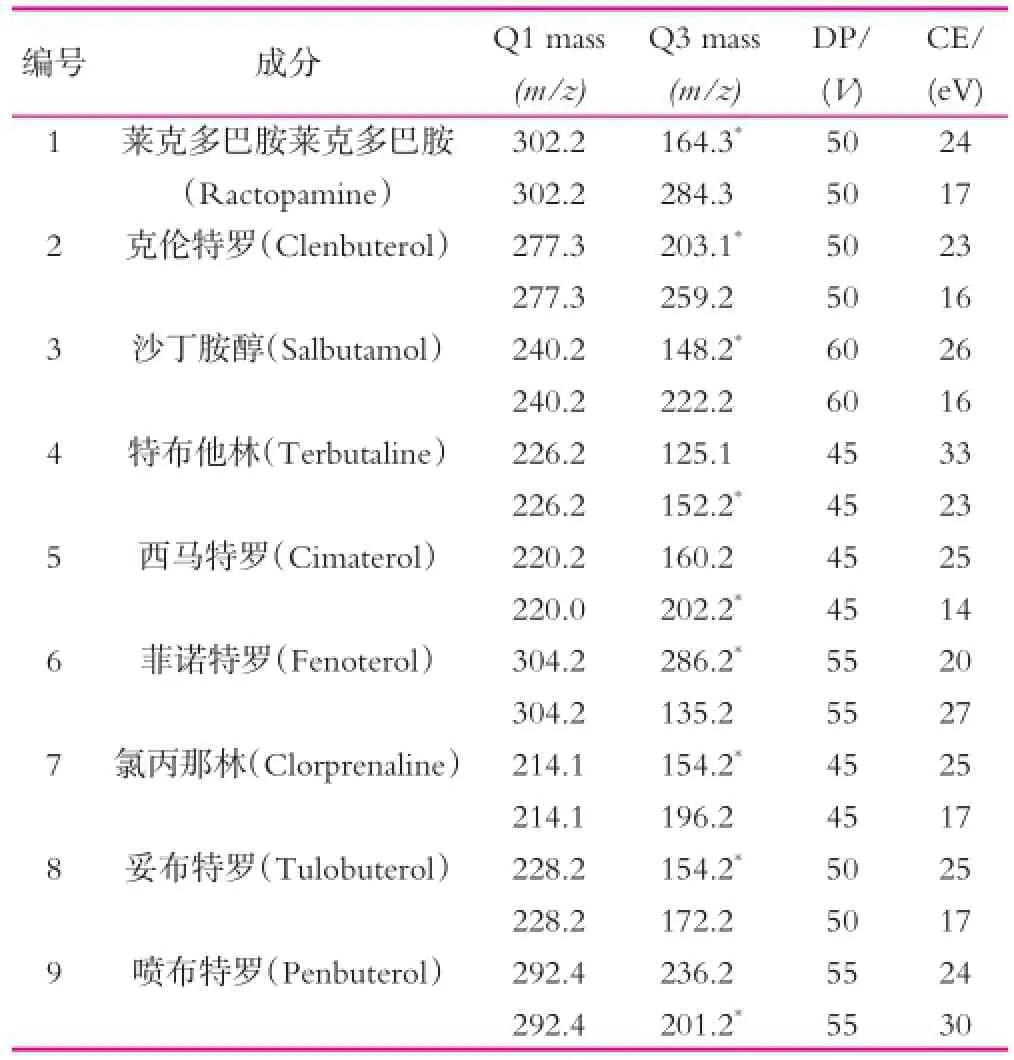
表1 9种β-受体激动剂的质谱优化条件
2.2提取条件优化
β2-受体激动剂属于强极性的化合物,含有胺类结构,部分还有1,2苯二酚(儿茶酚)等功能团[13]。该类药物的提取剂以甲醇、乙腈、丙酮、乙酸乙酯和二氯甲烷为主。由于液态乳中富含蛋白和脂质成分,且研究指出,乙腈具有良好的沉淀蛋白能力[14-15],因此本研究以乙腈为提取剂进行试验。比较了三氯乙酸、乙酸、氨水和EDTA-Mcllvaine缓冲液对提取效果的影响,结果表明EDTA-Mcllvaine缓冲液和三氯乙酸可提高乙腈的提取效果(图1)。EDTA为金属离子配合剂,可竞争配合β2-受体激动剂母核结构中的金属离子,使目标化合物游离出来。三氯乙酸是熊林等人针对传统酶法提取β2-受体激动剂改进的水解方法,避免了酶解耗时长、提取效果不稳定以及回收率偏低等缺点[4]。将EDTA和三氯乙酸同时添加到乙腈中,可将提取效果提高大约10%左右(图2)。因此本研究用2 mL的EDTA、8 mL乙腈和1 mL三氯乙酸作为提取剂进行后续试验。
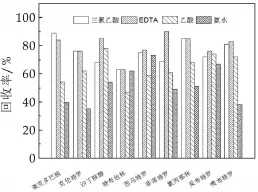
图1 不同物质对9种β2-受体激动剂回收率的影响
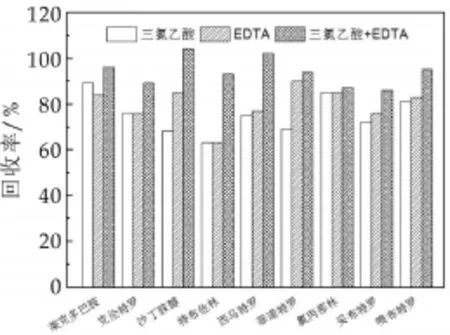
图2 EDTA-Mcllvaine和三氯乙酸对9种β2-受体激动剂回收率的影响
2.3脱水剂的优化
NaCl、MgSO4和Na2SO4是常用的脱水剂,液态乳中水分含量较高,因此分别用6 g NaCl,MgSO4,Na2SO4对上清液除水(图3)。结果表明,MgSO4作为脱水剂时回收率较低,这可能与目标物易与Mg2+形成螯合物有关。NaCl的效果最好,但提高用量后提取效果下降,其原因可能是部分目标物进入到水相中。
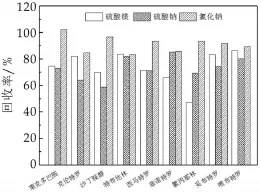
图3 脱水剂对9种β2-受体激动剂回收率的影响
2.4净化条件优化
液态乳经酸化乙腈和EDTA-Mcllvaine缓冲液提取后,已清除了蛋白质和大部分干扰物。为进一步提高净化效率,采用分散固相萃取法净化手段对样品进行处理。本研究选取C18,NH2,PSA比较其净化效果。结果表明,在相同用量下(100 mg),PSA作为净化剂的效果最好(图4)。三种填料均以硅胶为基质,分别键合C18、氨丙基和N-丙基乙二胺。PSA可有效去除提取液中的脂类和糖类物质,并且对β2-受体激动剂无显著吸附。液态乳中乳糖和乳脂肪含量较高,这可能是PSA具有较好净化作用的主要原因。
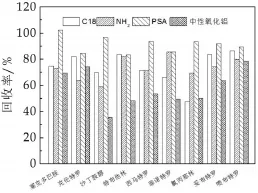
图4 净化剂对9种β2-受体激动剂药物回收率的影响
2.5基质标准曲线、线性范围和检出限及定量限
在质谱分析中,浸提物共流出组分对电喷雾离子化效果和检测信号有增强作用。为消除基质效应对β2-受体激动剂测定的影响,以经验证不含有9种β2-受体激动剂药物的空白液态乳提取液作为标准溶液的稀释液,采用优化后的前处理方法,制作基质加标工作曲线,得到线性回归方程的相关系数r2在0.991 ~0.998之间,线性范围为1.0~200 μg/kg。说明9种药物在该质量分数范围内线性关系良好。以定量离子信噪比(S/N)大于3为样品的检出限(LOD),以S/N大于10为定量限(LOQ),得到9种β2-受体激动剂的LOD为0.03 ~0.13 μg/kg,LOQ为0.1 ~0.4 μg/kg(见表2)。
2.6方法的回收率和精密度
采用标准添加法,在空白液态乳样品中分别添加3个水平的混合标准溶液进行回收率试验,每个添加水平依据本实验方法做6次平行测定,得到每个β-受体激动剂残留量的平均回收率为78.2~96.0%,相对标准偏差(RSD)为3.7~13.7%,符合兽药残留检测相关标准的要求。
3 结论
本文结合液态乳基质特性和β2-受体激动剂类兽药的理化性质,展开了QuEChERS–液相色谱法质谱法测定液态乳中9种β2-受体激动剂药残留的分析方法研究。对液态乳中9种β2-受体激动剂兽药残留的提取、盐析和净化过程中的各种影响因素进行了评价与优化,提高了提取方法效率。该方法前处理方法快速、简洁,定量方法准确,成本较低,能够满足对液态乳样品中β2-受体激动剂兽药残留的快速检测,也适用于大量筛查β-受体激动剂残留。
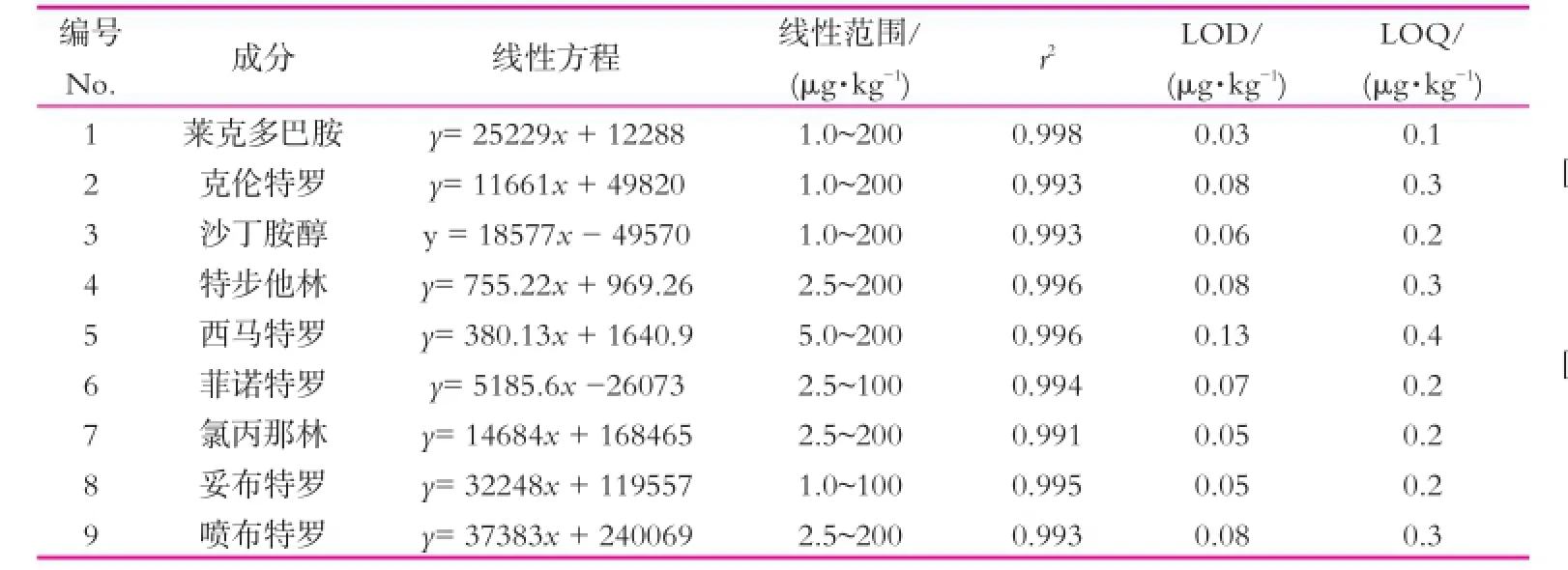
表2 9种β2-受体激动剂的线性方程、线性范围、相关系数(r2)、检出限(LOQ)和定量限(LOD)

表3 9种β2-受体激动剂的回收率和RSD
[1]JUAN C,IGUALADA C,MORAGUES F,et al.Development and validation of a liquid chromatography tandem mass spectrometry meth⁃od for the analysis ofβ-agonists in animal feed and drinking water[J]. J Chromatogr A,2010,1217:6061-6068.
[2]王培龙,范理,苏晓鸥,等.分子印迹固相萃取-气相色谱-质谱法测定猪尿中4种β-受体激动剂[J].分析化学,2012,40(3):470-473.
[3]苗虹,邹建宏,范赛,等.高效液相色谱-离子阱质谱法测定尿液中β2-受体激动剂及β-受体阻断剂[J].谱,2010,28(6):572-578.
[4]熊琳,李维红,高雅琴,等.肉品中β-受体激动剂类药物残留检测技术研究进展食品安全质量检测学报[J].2015,6(2):528-533.
[5]BLOMGREN A,BERGGREN C, HOLMBERG A.Extraction of clen⁃buterol from calf urine using a mo⁃lecularly imprinted polymer fol⁃lowed by quantitation by high-per⁃formance liquid chromatography with UV detection[J].Chromatog⁃raphy A,2002,975:157-164.
[6]吴平谷,王强,陈慧华,等.同时检测动物肌肉中26种β2-兴奋剂和激素残留[J].分析化学,2008,36(11):1476-1482.
[7]王炼,黎源倩.高效液相色谱法测定动物性食品中4种β2-兴奋剂[J].中国卫生检志,2008,18(7):1227-1230.
[8]ANASTASSIADES M,LEHOTAY S J,STAJNBAHER D,et al.Fast and easy multiresidue method employing acetonitrile extraction/parti⁃tioning and dispersive solid-phase extraction for the determination of pesticide residues in produce[J].Journal of Aoac International,2003,86 (2):412-431.
[9]李宁,林涛,邵金良,等.基质固相分散-超高效液相色谱-质谱检测器测定牛奶中9种类固醇激素残留[J].色谱,2015,33(11):1163-1168.
[10]郑小平,孟 瑾,何亚斌,等.QuEChERS-UPLC-MS/MS法同时测定羊奶中8种β-内酰胺类抗生素[J].食品与机械,2015,31(3):66-69.
[11]黎娟,乔庆东,庄景新,等.改进的高效液相色谱-串联质谱方法同时测定动物性食品中4种β2-受体激动剂残留[J].色谱,2016,34(2):170-175.
[12]蔡英华,薛毅,张,等.UPLC-MS/MS法测定动物源性食品中4个四环素类药物和10个β受体激动剂类药物残留[J].药物分析杂志,2014,34(7):1 223-1 230.
[13]罗辉泰,黄晓兰,吴惠勤,等.分散固相萃取-同位素稀释-高效液相色谱-串联质谱法同时测定猪肉中26种β-受体激动剂[J].色谱,2016,34(5):481-489.
[14]孙雷,张骊,汪霞,等.超高效液相色谱-串联质谱法对动物源性食品中13种β-内酰胺类药物残留的检测[J].分析测试学报,2009,285:576-580.
[15]郭萌萌,李兆新,谭志军,等.分散固相萃取/液相色谱-串联质谱法测定水产品中的8种青霉素残留[J].分析测试学报,2011,30(9):969-975.
Determination of 9β2-agonist drug residues by high performance liquid chro⁃matography-tandem mass spectrometry with QuEChERS in milk
LI Jing-yan,GUO Chun-feng,CUI Li-hui,LIU La-ping
(College of Food Science and Engineering,Northwest A&F University,Food Quality Supervision and Inspection Test⁃ing Center(Yangling),Ministry of Agriculture,Yangling 712100,China)
A method was developed for simultaneous determination ofβ2-agonist drug residues in milk by QuEChERS-high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS).The milk was extracted with acetonitrile,EDTA-Mcllvaine and tri⁃chloroacetic,dehydrated with NaCl,purified with PSA sorbent and separated by Shim-pack XR-ODS(3.0mmi.d×75mm)column using acetonitrile and water(containing 0.1%formic acid)as mobile phase.The mass spectrometric acquisitions were carried out by means of multi⁃ple reaction monitoring(MRM)in positive ionization mode.The linear relation of 9β2-agonist drug was good and the correlation coeffi⁃cient was more than 0.991.The limit of detection(LOD)of 9β2-agonist drugs was 0.03~0.13 μg/kg and the limit of quantification(LOQ) was 0.1~0.4 μg/kg.The recovery rates of the target analyte were between 78.2%~96.0%.The relative standard deviations(RSD)were less than 13.7%.The method was reliable for the determination ofβ2-agonist drug inmilk.
milk;β2-agonist drug residues;QuEChERS;HPLC-MS/MS
TS252.7
A
1001-2230(2016)11-0049-04
2016-06-22
李婧妍(1980-)女,实验师,从事食品和农产品中农药和兽药残留检测工作。
刘拉平
