光谱法鉴定手性化合物的绝对构型
——从仪器表征到理论计算
2016-02-09王娟杨笑迪
王娟杨笑迪
(1复旦大学化学系,上海200433;2复旦大学先进材料实验室,上海200438)
光谱法鉴定手性化合物的绝对构型
——从仪器表征到理论计算
王娟1杨笑迪2,*
(1复旦大学化学系,上海200433;2复旦大学先进材料实验室,上海200438)
手性化合物的结构确定,尤其是新型手性化合物的绝对构型测定一直是不对称研究的重要工作。除单晶测试外,光谱学方法近年来被广泛应用于手性分子结构鉴定,主要包括电子和振动圆二色谱、旋光光谱、旋光拉曼谱等。本文对上述测试方法的原理、应用范围和相关理论计算方法做了介绍。把谱学测试与理论计算相结合,将成为手性分子结构鉴定的重要发展方向。
绝对构型;理论与计算化学;电子圆二色谱;振动圆二色谱;旋光光谱
自1848年Pasteur从外消旋的酒石酸铵钠中分离出左旋与右旋的酒石酸铵钠晶体,到1874年J.H.van′t Hoff与J.A.Le Bel提出碳原子的四面体模型,立体化学便成为有机化学及药物化学家的重要研究课题之一[1]。从19世纪至今,人们对有机化学的认知发生了根本性转变,手性化合物已渗透到有机化学与药物化学的各个领域。尤其在药物研发过程中,药物分子的功能性与其立体构型密切相关。因此,准确有效地确定手性化合物的绝对构型至关重要。
手性化合物绝对构型的测定经历了漫长的探索与实践。最早确定分子手性的方法是有机合成法,即从初始已知手性的化合物开始,通过手性控制的有机化学反应,将其转化为目标化合物的方法。而有机反应路线过渡态计算,作为合成实验的有力补充,已经辅助有机化学工作者攻克了众多富有挑战性的复杂手性化合物[2]。从仪器表征角度出发,目前确定手性分子绝对构型的方法主要有X射线单晶衍射法、核磁共振(NMR)法和光谱学方法等。在各类测量方法中,X射线单晶衍射法作为一种可以精确测定分子三维空间结构的物理方法,是判断手性结构最有效的方法,其测试原理是基于分子中各原子对X射线的反常散射效应[3]。但由于该方法测试条件较为严苛,很多手性化合物产量低且单晶难以培养,因此仍然需要继续探索在溶液中测量手性分子绝对构型的方法。NMR方法将待测分子与手性试剂反应,通过化学位移的变化规律推断待测分子的绝对构型,该方法要求待测手性分子含有可与手性试剂发生反应的某些基团。与X射线单晶衍射法和NMR相比,光谱学方法对样品的要求不高,测试范围广泛,主要是基于旋光原理,通过旋光光谱、圆二色谱、旋光拉曼谱等方法测定化合物的结构信息,该类测试关键在于如何对测试谱图进行解析。以光谱学方法中的圆二色谱法为例,早期发展的构型判断规律有饱和环酮的“八区律”、Klyen的内酯扇形区规律、共轭双键和共轭不饱和酮的螺旋规律等[4]。分析数据时需要有类似化合物的参考文献作为数据支持,在判断不满足相关规律的新型手性化合物时存在难度。随着越来越多的新型手性化合物不断涌现,谱图指认与解析限制了该方法的应用。量子化学理论的发展和计算机技术的进步很好地解决了结构与谱图的对应问题[5,6]。将理论计算与仪器表征相结合,解决手性分子结构判定中遇到的各类问题,已经成为鉴定手性分子绝对构型的新趋势。本文旨在总结当前测定手性化合物结构的主要光谱学表征手段和辅助的理论计算方法,判定相对构型已知的手性化合物的绝对构型。
1 光谱学方法的测试原理与计算方法

图1 Franck-Condon原理示意图
光谱学方法是检测物质结构的重要手段,待测物对某些波长的电磁波产生的吸收或发射现象,可反映出待测物的结构信息。手性对映体在立体的结构上互为镜像,在光学性质上的差异主要表现在对偏振光的响应上。旋光性是最早识别对映异构体的方法,该方法用旋光的方向指示手性化合物的绝对构型。在光谱学方法中,圆二色谱法是应用最广泛的手性分子结构测定方法。当左圆偏振光与右圆偏振光通过手性化合物溶液时,左右圆偏振光的传播速率和吸收程度均发生变化。将摩尔吸光系数之差(Δε)随波长的变化作图可获得圆二色谱(circular dichroism,CD)。如果体系没有手性,则没有CD信号。根据手性化合物对平面偏振光的吸收机理差异,可以将圆二色谱方法分为电子圆二色谱(electronic circular dichroism,ECD)和振动圆二色谱(vibrational circular dichroism,VCD)。
在ECD测试中,手性化合物对平面偏振光的吸收是由电子吸收光子后产生电子能级之间的跃迁引起的,属于电子吸收光谱。VCD对应的吸收光谱为振动光谱,振动光谱是在同一电子能态下,不同振动能级之间的跃迁产生的。从Franck-Condon原理示意图(图1)可以清楚分辨电子能级与振动能级的区别。图中Q为分子的正则坐标,E为分子体系的能量。分子吸收光子后自基态S0跃迁至某一激发态Sn(图中仅画出第一个激发态S1),即对应着电子能级之间的跃迁。手性分子在某一电子态(以S0为例)下具有3n-6个振动模式,图中标出的0、1、2、……为S0的第j个振动模式对应的振动量子数。相邻振动能级间的能量差为hvj,vj是第j个振动模式的振动频率。在室温下,认为分子处于振动基态,主要对应0→1的跃迁。由于电子能级的能量间隔要大于振动能级,ECD所用的平面偏振光波长范围一般在200-400 nm,属于紫外区。VCD测试时设定的平面偏振波长在800 nm以上,为红外光谱范围。
1.1 电子圆二色谱
ECD法由于其干扰少,测试过程快捷简便,图谱包含信息量大,且发展总结了相应的图谱与构型对应的半经验规律,被广泛应用在手性化合物的测定上。随着量子化学理论的发展和计算机技术的进步,ECD谱可通过纯理论计算方法进行模拟,为通过ECD测量鉴定手性化合物的绝对构型提供了可行性[5]。
对于手性分子,从基态跃迁至第k个激发态对应的左右偏振光吸收系数之差Δεk由该跃迁对应的转动强度Rk决定[7]:

式(1)中,Δεk(λ)与波长λ呈函数关系,λk为对应的吸收峰的峰位。
转动强度与跃迁偶极矩和跃迁磁偶极矩有关,可表示为[8,9]:

其中Ψ0和Ψk表示基态和第k个激发态的波函数,u和m是电偶极矩算子和磁偶极矩算子,Im表示“虚部的”。根据式(2)可计算出各个激发态对应的转动强度。实际测试时的情况与式(1)描述的并不相符,式(1)描述的是某个特定波长λk下Rk与Δεk的相互关系。实际测量的是整个紫外吸收带的Δε。将波长转化为能量并通过高斯分布函数展宽[7],就得到Δε与能量E的关系:

式(3)中,ΔEk是基态跃迁至第k个激发态对应的跃迁能,σ是半峰宽。将通过量化计算得到的电子跃迁能和转动强度代入式(3)中,就可拟合出Δε与能量E的关系曲线,即ECD谱线。
具体的计算步骤主要包括:对已知相对构型的手性化合物建模并优化分子结构;对化合物进行激发态计算,获得ΔEk和Rk,通常采用含时密度泛函(TDDFT)方法进行计算;根据式(3)拟合ECD曲线,将拟合的谱图与实验测得的谱图进行对比,与实测值接近的即为化合物的正确构型。对于柔性分子体系,由于分子在溶液中存在多种构象,在结构优化前须进行构象搜索,将各构象的ECD谱按照该构象的分布概率进行加权,再与实测数据进行比较。
1.2 振动圆二色谱
与ECD相比,VCD的应用范围更加广泛。ECD要求手性化合物必须在紫外区有吸收峰,对于不含有生色团(紫外吸收)的手性化合物,VCD仍可进行圆二色性测试。由于振动光谱谱图的复杂性,VCD很难发展出经验的方法来解释结构与谱图的对应关系,这在很大程度上限制了VCD的应用范围。在量子化学计算的推动下,VCD谱图可通过纯理论计算来拟合,不再局限于依赖经验规律进行构型判断[6,10]。近年来VCD的应用越来越广泛,由于几乎所有的手性分子都能产生VCD谱图,VCD已成为鉴定手性化合物绝对构型的有力工具。
VCD描述的是手性化合物在红外光谱区左旋圆偏振光和右旋圆偏振光的吸收系数之差随波长的变化关系。因此VCD的计算步骤与ECD类似,差别在于VCD在理论计算时考查的是化合物的振动吸收,不涉及激发态信息。对于第j个正则模式对应的振动跃迁,对应的转动强度Rj定义为[10]:

其中Ψ0,0和Ψ0,j对应的都是基态下的波函数,在第j个正则模式下,自振动基态跃迁至第一个振动能级,对应的振动频率为vj。与ECD的情况类似,用转动强度来描述VCD谱线的强度[6]:

VCD谱通常用洛伦兹函数来展宽[11],根据Δε与振动频率v的关系,拟合出VCD谱线。

具体的计算步骤主要包括:对已知相对构型的手性化合物建模并优化分子结构;对化合物进行频率分析,获得vj和Rj,通常采用高斯软件包[12]中的DFT方法,运用关键词freq=VCD进行计算;根据式(6)拟合VCD曲线,将拟合的谱图与实验测得的谱图进行对比,与实测值接近的即为化合物的正确构型。对于柔性分子体系,仍须先进行构象搜索。
1.3 旋光光谱法
旋光性是最早识别对映异构体的方法。旋光现象是由于平面偏振光通过旋光性物质时,组成平面偏振光的左旋圆偏振光和右旋圆偏振光在介质中的传播速度不同,使平面偏振光的偏振面旋转了一定角度造成的。将比旋光度随入射偏振光波长的变化记录下来,就得到旋光色散曲线(optical rotatory dispersion,ORD)。
在判断化合物的立体结构时,无论是测量ORD谱还是CD谱,得到结论应当是一致的。通常CD谱的谱形更尖锐,峰位与紫外或红外光谱对应,比ORD谱便于分析。对于结构相近的手性化合物,由于吸收峰位十分接近,CD谱图形状相近,这时ORD谱可以提供更多信息。在条件允许时,最好是CD谱与ORD谱二者同时测试[13]。运用高斯软件包中的Polar=OptRot指令,可以直接计算指定波长下的旋光度。手性化合物的旋光度随偏振光的波长发生变化,在某特定波长下甚至为零值。因此,仅用某一波长的旋光性并不能完全判定化合物的绝对构型,通过ORD谱线判断手性分子的结构更具有说服性。
在波长λ处,比旋光度的理论表达式为[11]式中Vm和M分别对应摩尔体积和摩尔质量;φ(λ)定义为其中N为阿伏伽德罗常数,β(λ)为电子偶极-磁极极化率β0αβ的平均值[11,14]:

式(7)中,h为普朗克常数,c为光速,ωk为吸收峰位在λk的电子态跃迁(S0→Sk)对应的频率。一种更方便的表达方式是通过摩尔旋光度来描述ORD[15]:
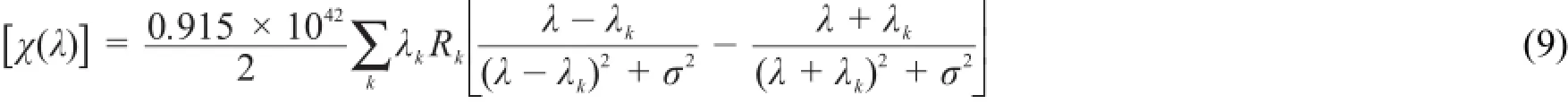
将拟合的ORD谱图与实验测得的谱图进行对比,与实测谱图接近的即为化合物的正确构型。
1.4 其他光谱学方法
1.4.1 圆二色谱激子手性法
圆二色谱激子手性方法(exciton chirality CD,ECCD)从谱图信号产生机理来分析,属于ECD范

由于式(8)在λ=λk时存在奇点,式(9)是更普适的可用于共振区和非共振区的ORD计算公式[11,15]:畴。上世纪50年代末,ECCD方法逐渐形成。Harada和Nakanishi[16]对ECCD法进行了大量的系统研究,ECCD方法得以大量推广,成为测定有机分子绝对构型的重要方法。
该方法的主要机理为:当分子中的手性位置含有两个相同或波长接近的生色团时,经光照激发后,两个发色团激发态之间发生激子耦合,此时激发态分裂成两个能级,形成两个符号相反的Cotton效应。当确定了生色团跃迁偶极矩的方向,根据Cotton效应的符号就可以确定两个发色团在空间的绝对立体化学。ECCD法也适用于具有三个或三个以上发色团的化合物。随着ECCD法的不断深入研究,各类合适的生色团通过衍生化引入到手性化合物中[17,18],其应用范围不断扩大,越来越多的应用于各种天然产物和合成有机化合物的绝对构型确定。
1.4.2 旋光拉曼光谱法
旋光拉曼光谱(raman optical activity,ROA)是对手性分子绝对构型进行研究的新型光谱技术。ROA被认为是继X射线晶体衍射和核磁共振技术之后,研究手性分子的又一重大进展。手性化合物的拉曼光学活性在20世纪70年代被预测并观测到[19]。手性分子的某一个振动模式对于左右圆偏振光具有不同的散射截面,其差别量仅有散射截面的10-4,实验技术的局限限制了ROA的发展。近十年来伴随着理论和实验的进展,ROA的研究工作逐渐开展。人们通过第一性原理预测了手性分子的ROA谱图[20,21],通过理论预测与实验结果的对比,使得通过ROA确定手性分子的绝对构型成为可能。
ROA的机理源于电偶极矩、磁偶极矩和电四偶极矩之间的耦合,其中的磁性来源于分子振动时产生的电荷流动所引起的磁场。ROA所测量的是比经过偶极矩过程的光吸收、光的散射更深的层次,涉及的是分子内跃迁的磁过程和四极矩过程;反映的是传统红外和常规拉曼技术无法提供的、更高层次的分子内部立体结构信息。目前理论层面的ROA计算主要是针对分子的电偶极-电偶极相互作用[22,23]和电偶极-电四偶极相互作用[24]。电偶极-磁偶极相互作用的计算机制仍有待完善[11]。
2 光谱学方法计算实例
前面对利用计算化学方法预测各类光学谱线的基本原理已经做了较系统的阐述。根据理论计算得到的谱图与实验测试获得的谱图进行比较从而确定化合物的绝对构型。构象搜索通常用分子力学方法来实现,常用的构象搜索软件包括MacroModel、HyperChem、Insight II、Spartan等。分子构象优化与参数计算则基于量子化学理论来获得。早期的量子化学方法主要是通过分子轨道理论求解薛定谔方程获得波函数和分子相关信息,如HF、CI、MP、半经验计算方法等。自20世纪70年代,密度泛函理论(DFT)得到了蓬勃发展。DFT以密度分布函数代替电子的波函数,因其明显的优越性受到广泛推广。近年来,多种手性化合物使用光谱学方法测定绝对构型。Stephens[10]、Frisch[22]、Berova[5,18,25]、Nafie[6]和Polavarapu[13,15,20,21]等科研工作者做了大量的谱学计算与预测工作。下面列举一些通过光谱学方法判断手性化合物绝对构型的实例。
2.1 (4S,5R,6R)-oxysporone的绝对构型确定[25]
(4S,5R,6R)-oxysporone(化合物1,如图2所示)是自尖刀镰孢菌中分离出的天然产物,目前并无X射线单晶衍射数据证明化合物1的绝对构型,其相对构型是通过NMR测试确定的。
Mazzeo等[25]首先使用分子力学方法对化合物1的对映异构体分别进行构象搜索,获得6种优势构象。将这些构象在DFT/B3LYP/cc-pVTZ水平上做结构优化,确定各个构象的分布率。分别在EtOH和CHCl3溶液中,测试了化合物1的ORD谱(图3(a)),ORD谱较为平坦且呈正值。基于优化出的几何结构,在TDDFT/B3LYP/aug-cc-pVDZ水平上对化合物1进行ORD计算,并按照分构象分布率进行加和得到理论预测谱图。发现(4S,5R,6R)-1的ORD计算结果与实测结果更接近,(4R,5S,6S)-1的谱线与实验测得的[α]绝对值相近,但ORD谱线与实测谱线互为镜像。这表明(4S,5R,6R)-1更“符合”化合物1的绝对构型。

图2 化合物1的指定构型(4S,5R,6R)-1(左)和它的反型(4R,5S,6S)-1(右)
ECD谱(图3(b))是在MeCN(8.6×10-3mol∙L-1)溶液中测量的。测量的数据显示:化合物1在230 nm处有一低振幅、负Cotton效应(Cotton effect,CE)峰,在200 nm处对应着一个高强度的正CE峰。ECD是在TDDFT/CAM-B3LYP/aug-cc-pVDZ水平上计算的,计算了前30个激发态的信息,半峰宽设定为0.4 eV。将理论计算数据与实验测量数据作对比,发现(4S,5R,6R)-1的ECD谱线与实验值的符号和趋势均十分接近,进一步证明了化合物的绝对构型为(4S,5R,6R)-1。
VCD计算是在B3LYP/6-31G(d)水平上,对各构象分别计算并按照其分布率加和获得的。VCD谱图(图4)中,各个峰的位置与在同一计算水平下预测的IR谱的峰位是一致的,并可通过频率计算将IR谱的各个吸收峰与振动模式作对照分析。(4R,5S,6S)-1和它的反型(4S,5R,6R)-1,它们的理论预测VCD谱图峰位一致,符号相反。VCD谱的分析结果与ORD和ECD是一致的。再一次确定了化合物1的绝对构型是(4S,5R,6R)-1。
2.2 α-benzylamino-coumarins的绝对构型确定[26]
α-benzylamino-coumarins(化合物2)可通过Mannich反应合成,产率高达99%,对映选择性达到83%ee。但其单晶难以培养,无法直接通过单晶衍射测试判断绝对构型。其衍生物培养出的单晶结构经X射线衍射测试后,发现晶胞中R和S构型交替排列,无法通过衍生物的相对构型反推化合物2的绝对构型。故采用ECD测试并结合理论预测的方法来确定手性化合物的绝对构型。针对R和S型的化合物2,首先使用Confab程序进行构象搜索,对能量间隔在0-83.7 kJ∙mol-1的几何结构做计算水平为B97D/6-31G*的结构优化并按照能量高低排序。以12.6 kJ∙mol-1为能量截点对剩余的分子构象做DFT/B3LYP/TZVP结构优化,并考虑PCM溶剂效应,获得各个构象的Boltzmann分布和分子几何。激发态信息采用B3LYP泛函,并选择不同的基组如6-31G*、TZVP、cc-pVDZ、6-311+G**进行比较计算。理论预测表明S型的ECD谱其峰位和符号与实验测试谱图完全吻合(图5),R型的ECD谱图与S型峰位一致,符号相反。因此化合物2的绝对构型为S型。
值得注意的是,文献中报导的多为有机小分子体系,这与当前的计算化学水平密切相关。对于单键较多、柔性较高的大尺寸分子,无论是判断其低能态优势构型还是计算分子的光谱性质,对理论化学的计算精度和计算机的计算能力都有较高要求。

图3 理论预测的(4S,5R,6R)-1和(4R,5S,6S)-1的ORD谱(a)与ECD谱(b)分别与实验测试的谱线对照分析

图4 实验测试的化合物1的VCD谱图(b),以及理论计算获得的(4R,5S,6S)-1 VCD谱(a)和(4S,5R,6R)-1 VCD谱(c)

图5 化合物2的理论计算与实验测试ECD谱图
3 总结与展望
与X射线单晶衍射法和NMR方法相比,光谱学方法在溶液状态下就可以测量手性分子的结构信息。在已知分子相对构型的前提下,将光谱学测试与理论计算相结合,发展计算分子谱学性质的理论方法,通过构建分子结构与谱图的对应关系,可确定手性分子的绝对构型。伴随着量子化学的快速发展,理论计算作为测试分析的有效辅助手段,其应用范围必将不断扩大。
鉴定手性化合物绝对构型的关键在于了解各类方法的原理和适用范围,选择最适合的测试手段来解决构型问题。当一种方法不足以判断分子的立体结构时,从科学研究的严谨性出发,应同时使用多种表征手段来收集化合物的结构信息。将多种仪器表征技术相融合,将分析测试与理论计算相融合,是手性分子结构鉴定的重要发展方向。
[1]林国强,孙兴文,陈耀全,李月明,陈新滋.手性合成——不对称反应及其应用.北京:科学出版社,2013:1-3.
[2]Cheong,P.H.Y.;Legault,C.Y.;Um,J.M.;Celebi-Olcum,N.;Houk,K.N.Chem.Rev.2011,111(8),5042.
[3]Bijvoet,J.M.;Peerdeman,A.F.;van Bommel,A.J.Nature 1951,168(4268),271.
[4]Lightner,D.A.;Gurst,J.E.Organic Conformational Analysis and Stereochemistry from Circular Dichroism Spectroscopy;Wiley-VCH: New York,2000.
[5]Berova,N.;Di Bari,L.;Pescitelli,G.Chem.Soc.Rev.2007,36(6),914.
[6]Freedman,T.B.;Cao,X.L.;Dukor,R.K.;Nafie,L.A.Chirality 2003,15(9),743.
[7]Diedrich,C.;Grimme,S.J.Phys.Chem.A 2003,107(14),2524.
[8]Bak,K.L.;Hansen,A.E.;Ruud,K.;Helgaker,T.;Olsen,J.;Jorgensen,P.Theor.Chim.Acta 1995,90(5-6),441.
[9]Grimme,S.Chem.Phys.Lett.1996,259(1-2),128.
[10]Stephens,P.J.;Devlin,F.J.;Pan,J.J.Chirality 2008,20(5),634.
[11]Polavarapu,P.L.Chem.Rec.2007,7(2),125.
[12]Frisch,M.J.T.;Schlegel,G.W.;Scuseria,H.B.;Robb,G.E.;Cheeseman,M.A.;Montgomery,J.R.;et al.Gaussian 09,Revision B.01; Gaussion Inc.:Wallingtord,CT,2009.
[13]Polavarapu,P.L.Chirality 2008,20(5),664.
[14]Giorgio,E.;Viglione,R.G.;Zanasi,R.;Rosini,C.J.Am.Chem.Soc.2004,126(40),12968.
[15]Polavarapu,P.L.Chirality 2006,18(5),348.
[16]Harada,N.;Nakanishi,K.Acc.Chem.Res.1972,5(8),257.
[17]Gawronski,J.;Skowronek,P.Curr.Org.Chem.2004,8(1),65.
[18]Tanaka,K.;Pescitelli,G.;Nakanishi,K.;Berova,N.Monatsh.Chem.2005,136(3),367.
[19]Barron,L.D.;Buckingham,A.D.Mol.Phys.1971,20(6),1111.
[20]Polavarapu,P.L.J.Phys.Chem.1990,94(21),8106.
[21]Polavarapu,P.L.;Hecht,L.;Barron,L.D.J.Phys.Chem.1993,97(9),1793.
[22]Frisch,M.J.;Yamaguchi,Y.;Gaw,J.F.;Schaeffer II,H.F.;Binkley,J.S.J.Chem.Phys.1986,84(1),531.
[23]Quinet,O.;Champagne,B.J.Chem.Phys.2001,115(14),6293.
[24]Quinet,O.;Liegeois,V.;Champagne,B.J.Chem.Theory Comput.2005,1(3),444.
[25]Mazzeo,G.;Santoro,E.;Andol,A.;Cimmino,A.;Troselj,P.;Petrovic,A.G.;Superchi,S.;Evidente,A.;Berova,N.J.Nat.Prod.2013, 76(4),588.
[26]Lin,H.;Tan,Y.;Wu,J.M.;Yang,X.D.;Chen,J.H.;Sun,X.W.Tetrahedron Lett.2015,56(7),913.
Determination of Absolute Configuration of Chiral Compounds Based on Chiroptical Spectroscopic Methods:From Instrument Characterization to Computational Chemistry
WANG Juan1YANG Xiao-Di2,*
(1Department of Chemistry,Fudan University,Shanghai 200433,P.R.China;2Laboratory of Advanced Materials,Fudan University,Shanghai 200438,P.R.China)
The determination of the absolute configuration(AC)of chiral compounds,especially the new chiral compounds,is one of the difficulties of the structure determination.The most widely used AC determination chiroptical spectroscopic methods include electronic and vibrational circular dichroism, optical rotatory dispersion,Raman optical activity etc.This mini review summarizes the main test principles, applications and corresponding theoretical calculation methods.With the developments of theoretical chemistry,the AC determination through instrumental characterization technique coupled with quantum chemical calculation,is envisaged to be the new trends ofAC assignment.
Absolute configuration;Theoretical and computational chemistry;Electronic circular dichroism;Vibrational circular dichroism;Optical rotatory dispersion
G64;O6
*通讯作者,Email:yangxiaodi@fudan.edu.cn
国家自然科学基金(21103023)
10.3866/PKU.DXHX201603022
www.dxhx.pku.edu.cn
