固相萃取/UPLC-MS/MS法检测饲料中18种喹诺酮类药物的研究
2016-02-07彭玉芬蔡杰蔡勤仁薛良辰黄晶凌芸辉
彭玉芬,蔡杰,蔡勤仁,薛良辰,黄晶,凌芸辉
( 珠海出入境检验检疫局技术中心,广东珠海519015)
固相萃取/UPLC-MS/MS法检测饲料中18种喹诺酮类药物的研究
彭玉芬,蔡杰,蔡勤仁,薛良辰,黄晶,凌芸辉
( 珠海出入境检验检疫局技术中心,广东珠海519015)
为了建立超高效液相色谱-串联质谱法测定饲料中18种喹诺酮类药物的分析方法,以0.1%乙酸乙腈提取饲料样品中的待测物,HLB柱净化,超高效液相色谱-串联质谱法测定,氘代同位素内标法定量。18种喹诺酮类药物在线性范围为0~1.0 mg/L,线性关系良好,相关系数分别在0.9989~0.9999之间,方法的检测限为0.005~0.2 mg/kg。在不同饲料中添加浓度范围为0.1~5.0 mg/kg,平均回收率为43.0%~101%,相对标准偏差小于11.2%。结果表明,该方法灵敏、高效、抗干扰力强,适用于饲料中的18种喹诺酮类药物的测定。
超高效液相-串联质谱法;固相萃取;饲料;喹诺酮类药物
喹诺酮类(quinolones)为人工合成的抗菌药,是一类人畜通用的药物。因其具有抗菌谱广、抗菌活性强、与其他抗菌药物无交叉耐药性和毒副作用小等特点,被广泛应用于畜牧、水产等养殖业中,包括在鸡、鸭、鹅、猪、牛、羊、鱼、虾、蟹等的养殖中用于疾病防治。饲料是动物的直接食品,它的安全与否直接关系到动物食品的安全,饲料的不安全因素会通过食物链对人类的安全健康造成危害。若长期食用被此类药物污染的食品,可能会诱导人类病原微生物产生耐药性和抗药性,严重时,会对食用者产生消化系统及中枢神经系统等不良反应[1]。欧盟、世界卫生组织及我国都对动物源性食品中喹诺酮类药物制定了最大残留限量,农业部也对饲料中喹诺酮类药物的使用作了相关规定[2]。因此,建立一种同时检测饲料中多种喹诺酮类药物的方法具有重要意义。
目前,饲料中喹诺酮类药物的检测方法不能覆盖常用的喹诺酮类药物,不利于养殖中全面监控,文献报道关于饲料中喹诺酮药物含量检测方法较少,主要有高效液相色谱法[3-4]、毛细管电泳法[5]和液质联用法[6-7]。饲料中18种喹诺酮类药物检测采用高效液相色谱法基质干扰大;毛细管电泳法难以普及。所以本研究采用超高效液相色谱-串联质谱法进行分析检测,该方法灵敏、高效、重现性好、杂质干扰小,满足饲料中喹诺酮药物检测的准确定性和定量的要求。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器 超高效液相色谱-串联API4000Q Trap 质谱仪(美国ABI公司);Hitachi CR22GH离心机(日本日立公司);ZymarkTurboVap@LV氮气浓缩仪(美国Caliper公司);KQ-500DE型超声波清洗仪(昆山市超声波仪器有限公司);负压固相萃取装置(美国Agilent公司);HS250B 型摇床(德国IKA 集团);MS3 basic 涡旋混合器(德国IKA 集团);Milli-Q超纯水系统(美国Millipore公司)。
1.1.2 药品与试剂 恩诺沙星、环丙沙星、沙拉沙星、诺氟沙星、氧氟沙星、培氟沙星、洛美沙星、二氟沙星、单诺沙星、麻保沙星、恶喹酸、氟甲喹、萘啶酸、司帕沙星、奥比沙星、氟罗沙星、依诺沙星、吡哌酸均、盐酸恩诺沙星-D5、盐酸环丙沙星-D8、诺氟沙星-D5和恶喹酸-D5(德国Dr.Ehrenstorfer公司),纯度大于95%;甲醇、乙腈和甲酸均为色谱纯(美国Merck公司);乙酸铵(色谱纯,美国Fisher Chemical公司);氨水(分析纯,广州化学试剂厂);Waters Oasis HLB固相萃取柱(3 mL /60 mg),使用前依次用3 mL甲醇,3 mL水活化。
标准物质储备液:分别称取18种喹诺酮类标准品适量,以甲醇溶解并定容至25 mL,4 ℃冰箱中避光保存。
混合标准工作液(10.0 mg/L):分别取18种喹诺酮类药物储备液适量于25 mL容量瓶中,用甲醇定容,4 ℃冰箱中避光保存。
4种喹诺酮类内标物质储备液:分别准确称取盐酸恩诺沙星-D5、盐酸环丙沙星-D8、诺氟沙星-D5和恶喹酸-D5标准品适量,以色谱纯甲醇溶解并定容至25 mL,4 ℃冰箱中避光保存。
内标混合标准工作液(10.0 mg/L):分别取4种喹诺酮类内标物质储备液适量于25 mL容量瓶中,用甲醇定容,4 ℃冰箱中避光保存。
5.0 mmol/L乙酸铵溶液(含0.1%的甲酸):溶解0.385 g乙酸铵于1000 mL水中,再加入1.0 mL的甲酸,4 ℃冰箱中保存。
1.2 样品前处理方法 称取2 g(精确到0.01 g)样品于50 mL聚丙烯离心管中,加入20 μL内标混合标准工作液,4 mL水浸泡1 h,加入20 mL 1 %乙酸乙腈,振荡15 min,超声波震荡提取10 min,4000 r/min离心5 min,上清液倒出至另一离心管,用乙腈定容至25 mL,取5 mL 50 ℃氮气浓缩至0.5 mL,加水至10 mL,混合均匀,备用。
移取上述样品液至Oasis HLB固相萃取柱净化,控制流速以不超过1.0 mL /min的速度通过固相萃取小柱,用6 mL水和6 mL 20 % 甲醇淋洗,抽干。6 mL 5%氨水-甲醇洗脱,收集洗脱液,50 ℃氮气浓缩至近干,加入2 mL 5 mmol /L 乙酸铵(含0.1% 的甲酸)-乙腈(90+10,V/V)溶解残渣,涡旋混匀,过0.22 μm微孔滤膜,用于LC-MS/MS检测。
1.3 仪器条件
1.3.1 液相色谱条件 色谱柱:UPLC ACQUITY BEH C18柱(2.1 mm× 100 mm,1.7 μm);柱温:35 ℃;进样量:5 μL;流动相A 为5 mmol /L 乙酸铵(含0.1% 的甲酸),B 为乙腈,梯度洗脱(表1)。

表1 梯度洗脱程序
1.3.2 质谱条件 离子源: 电喷雾离子源( ESI);正离子扫描;多反应监测(MRM)模式;喷雾电压:5500 V;雾化气:60 psi,气帘气:25 psi,辅助加热气:65 psi;碰撞气:Medium,四种气体均为氮气;离子源温度:550℃;碰撞能量、去簇电压、定性离子对及定量离子对等见表2。

表2 18种喹诺酮类药物的质谱分析优化参数
*代表定量离子
2 结 果
2.1 基质标准曲线及检测限 将混合标准工作液依次稀释得到标准系列,内标浓度0.05 mg/L,由低到高的浓度进行测定,以标准溶液浓度(X)为横坐标,以峰面积(Y)为纵坐标绘制标准曲线。由于样品基质溶液对18种喹诺酮类药物的灵敏度有不同程度的影响,试验用空白基质溶液稀释标准系列,消除基质效应。其中,线性范围为0~0.2 mg/L的标准系列是0、0.01、0.02、0.05、0.1、0.2 mg/L,0~1.0 mg/L的标准系列是0、0.05、0.1、0.2、0.5、1.0 mg/L。检测限依据信噪比S/N>3确定。回归方程和检测限见表3-表5,从表中可以看出,在建立的色谱质谱条件下,18种喹诺酮类药物在各自的线性范围内呈现良好线的线性关系。
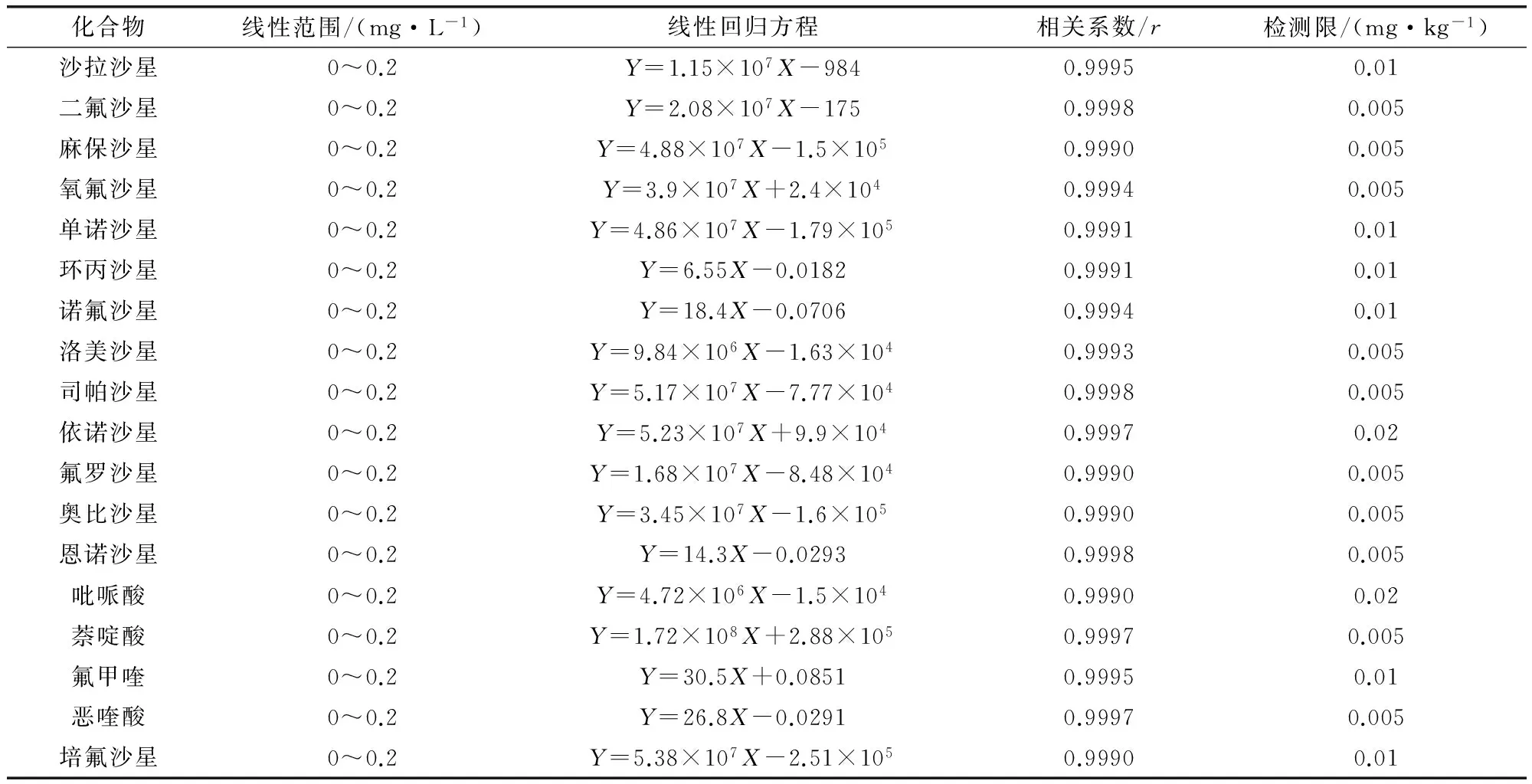
表3 18种喹诺酮类药物在全价配合饲料中的线性回归方程、相关系数和检测限

表4 18种喹诺酮类药物在浓缩饲料中的线性回归方程、相关系数和检测限

表5 18种喹诺酮类药物在预混饲料中的线性回归方程、相关系数和检测限
2.2 回收率及精密度 在空白配合饲料、浓缩饲料和预混饲料中添加18种喹诺酮类药物混合标准工作液和内标混合标准工作液,配合饲料0.1、0.5、1.0 mg/kg,浓缩饲料和预混饲料0.1、0.5、1.0、5.0 mg/kg,内标浓度为0.5 mg/kg,每个浓度6个重复,同时做空白对照,按1.2处理方法进行处理后上机测试(图1)。回收率和精密度见表6,从表6中可以看出,18种喹诺酮类药物平均回收率在43.0%~101%之间,相对标准偏差4.0%~11.2%之间,说明该法在不同饲料中准确度和精密度好,稳定可靠。

表6 18种喹诺酮类药物在饲料中回收率及相对标准偏差(n=6)

续表

续表

图1 配合饲料添加18种喹诺酮类药物0.1 mg/kgTIC图
3 讨论与小结
3.1 提取溶剂的选择 饲料是由能量饲料(谷物等)、蛋白质饲料(鱼粉、骨肉粉等)、矿物质饲料(贝壳粉、食盐等)以及各种添加剂(微量元素、维生素等)经过加工混合而成。饲料按营养成分分为配合饲料、浓缩饲料和添加剂预混合饲料等,饲料基质复杂且干燥,喹诺酮药物在饲料中不易解离,直接用有机溶剂提取会造成很大的损失,回收率也受到影响,所以先用两倍的水浸泡。喹诺酮类药物属于酸碱两性化合物,在多数溶剂中溶解性差,在pH≤4或pH≥9时,喹诺酮类药物易溶于含水相,pH 6~8时易于有机溶剂反萃取,绝大多数样品使用酸化或碱化溶剂作为提取溶剂[8],试验比较了乙腈-2%氢氧化氨溶液、1%乙酸-乙腈溶液、1%三氯乙酸-甲醇溶液和N,N-二甲基甲酰胺-0.1 mol/L盐酸溶液提取效率,发现1%乙酸乙腈作为提取溶剂回收率较高且杂质干扰最小,故本实验选用1%乙酸乙腈溶液作为提取溶剂。3.2 净化条件的确定 饲料基质复杂,浓缩后直接检测干扰大且污染仪器。参考了GB/T 23412-2009[9]、GB/T 21312-2007[10]、SN/T 3649-2013[11]和文献[12-13]的净化方法,比较正己烷、Waters OasisMCX阳离子交换固相萃取小柱、PAX阴离子交换固相萃取小柱以及Oasis HLB固相萃取小柱净化效果,结果发现用Oasis HLB固相萃取小柱净化,20%甲醇水淋洗,6 mL 5%氨水-甲醇洗脱,18种喹诺酮类药物回收率最理想。
3.3 质谱条件和色谱条件的选择 结果表明,喹诺酮类化合物的二级质谱中主要的碎片离子峰是喹诺酮药物分子的脱水峰[M+H-18]+、脱羧峰[M+H-44]+以及脱羧后哌嗪环断裂发生结构重排失去C2H4NR2的产物离子[M+H-CO2-C2H4NR2]+。较低丰度的离子峰有[M+H-64]+、[M+H-CO2-C2H4NR2-14]+,[M+H-64]+应是氟喹诺酮脱羧后再丢失HF分子产生的带电离子,[M+H-CO2-C2H4NR2-14]+是氟喹诺酮脱羧后哌嗪环断裂发生结构重排失去C3H6NR2的产物离子。依据食品法典委员会和欧盟第657/2002/EEC号中有关规定[13],选择最强的两对离子对作为定量和定性离子对。
流动相的组成和配比会影响待测物的色谱行为及其灵敏度,试验选择含0.1% 的甲酸的5 mmol/L乙酸铵与乙腈作为流动相,能较好的分离18种待测物和消除拖尾现象。
研究采用同位素内标液质联用技术测定饲料中18种喹诺酮类药物,方法操作简单、快速、回收率和精密度好,覆盖了常用的喹诺酮类药物,应用该方法,对33份饲料中的喹诺酮类药物进行了检测,7份饲料中检出恩诺沙星残留,含量在0.2 ~ 1.4 mg/kg之间,为饲料中喹诺酮类药物溯源和卫生监督提供了技术依据。
[1] 张俊丰,陈琳. 氟喹诺酮药物在兽医临床的应用[J]. 兽药与饲料添加剂,2002,7(5):13-15.
[2] 中华人民共和国农业部. 饲料药物添加剂使用规范(农业部公告第168号)[EB/OL].
[3] 贾涛. 液相色谱-荧光法测定饲料中氟喹诺酮药物[J]. 检测分析,2012,12:43-46.
[4] 单乃荣,何琼,余莲,等. 液相色谱法测定饲料中氟喹诺酮类含量方法研究[J]. 广西农学报, 2012, 27(4):51-55.
[5] 李慧,祁克宗,邵黎,等. 高效毛细管电泳用于饲料中5种氟喹诺酮类药物的同时测定[J]. 中国饲料,2009,20:33-35.
[6] 梁君妮,刘宁,曹鹏,等. HPLC-MS/MS测定饲料中16种氟喹诺酮类药物残留量[J]. 食品研究与开发,2013,34 (22):39-42.
[7] 彭丽,吴宁鹏,张发旺,等. 液相色谱-串联质谱法测定饲料中磺胺类和喹诺酮类药物的含量[J]. 中国兽药杂志,48(10):53-59.
[8] 李俊锁,邱月明,王超. 兽药残留分析[M]. 上海:上海科学技术出版社,2002, 285-287.
[9] 中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会. 蜂蜜中19种喹诺酮类药物残留量的测定方法液相色谱-质谱/质谱法.GB/T 23412-2009[S].
[10]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.动物源性食品中14种喹诺酮药物残留检测方法液相色谱-质谱/质谱法. GB/T 21312-2007[S].
[11]中华人民共和国国家质量监督检验检疫总局.饲料中氟喹诺酮类药物含量的检测方法液相色谱-质谱/质谱法. SN/T 3649-2013[S].
[12]Jonas A R, Felix G R, Susanne R. Quantitation and identity confirmation of residues of quinolones in tilapia fillets by LC-ESI-MS-MS QToF[J]. Anal Bioanal Chem, 2009, 394: 2213-2221.
[13]王金秋,马建民,夏曦,等. 超高效液相色谱-串联质谱法同时测定猪肌肉中13种喹诺酮药物残留[J]. 质谱学报,2014,35(2):185-192.
[14]European Union(2002/657/EEC), Official Journal of the European Union[Z], L221/8-9, 2002, EN.
(编辑:陈希)
Determination of 18 Quinolones in Feeds with Solid Phase Extraction by UPLC-MS/MS
PENG Yu-fen, CAI Jie, CAI Qin-ren, XUE Liang-chen, HUANG Jing, LING Yun-hui
(ZhuhaiInspection&QuarantineBureauTechnologyCenter,Zhuhai,Guangdong519015,China)
An ultra high performance liquid chromatography - tandem mass spectrometry (UPLC-MS/MS) mehtod was developed to determine 18 quinolones in feeds. 18 quinolones were extracted by 0.1% acetic acid-acetonitrile solution , cleaned up with SPE of HLB column and detected by UPLC-MS/MS. Quantitative results were based by isotope internal standard method. The calibration curve of 18 quinolones showed good linearity in the range of 0~1.0 mg/L with conrrelation coefficient of 0.9989 and 0.9998 respectively. The detection limits of method were 0.005~0.2 mg /kg. The average recoveries of 18 quinolones at the spiked levels of 0.1~5.0 mg/kg in feeds ranged from 43.0%~101% with relative standard deviation less than 11.2%. Experiments showed that the method was sensitive, high efficiency and good anti-interference. The method adapted to the detection of 18 quinolones in feeds.
UPLC-MS/MS; solid phase extraction; feeds; quinolones
珠海出入境检验检疫局科技计划项目( ZH 2014-3)
2016-08-05
A
1002-1280 (2016) 12-0051-08
S859.79
