SGLT2抑制剂——3-羰基达格列净的合成新方法*
2016-01-17姜琳琳徐为人汤立达汪文锦赵桂龙济宁学院化学与化工系山东曲阜7355天津药物研究院天津市新药设计与发现重点实验室天津30093
姜琳琳,尹 玲,徐为人,汤立达,汪文锦,赵桂龙(.济宁学院化学与化工系,山东曲阜 7355;.天津药物研究院天津市新药设计与发现重点实验室,天津30093)
SGLT2抑制剂——3-羰基达格列净的合成新方法*
姜琳琳1,尹玲1,徐为人2,汤立达2,汪文锦2,赵桂龙2
(1.济宁学院化学与化工系,山东曲阜273155;
2.天津药物研究院天津市新药设计与发现重点实验室,天津300193)
摘要:以4-甲氧基苯基-β-D-吡喃葡萄糖苷为原料,经9步反应制得新型关键中间体2,4,6-三-O-苄基-3-羰基-D-葡萄糖酸内酯乙二硫醇缩酮(10); 10与5-溴-2-氯-4'-乙氧基二苯甲烷经亲核加成反应制得甲基糖苷(12); 12在BF3·Et2O催化下经Et3SiH还原、AlCl3脱去苄基、再经PhI(CF3CO2)2氧化脱除乙二硫醇保护基合成了SGLT2抑制剂3-羰基达格列净,共13步反应,总收率9%,该合成路线包含了11个新化合物的合成,其结构经1H NMR,13C NMR,IR和HR-ESI-MS表征。
关键词:SGLT 2抑制剂;达格列净; 3-羰基达格列净;合成方法
技术研究计划(14JCQNJC12900,14JCZDJC33500)
通信联系人:赵桂龙,副研究员,E-mail:zhao_guilong@126.com;汪文锦,硕士,E-mail:wangwenjins@163.com
2型糖尿病是一种以高血糖为主要特征的慢性代谢性疾病,治疗不及时或者不充分会引起严重的并发症。尽管目前临床上有多种降血糖药物,但是由于药物抵抗等原因,全新作用机制的降血糖药物在临床上仍然有迫切需求。Na+依赖性葡萄糖共转运体2(SGLT2)是近几年出现的2型糖尿病治疗的新靶点[1-2],目前已经有6个SGLT2抑制剂药物(Dapagliflozin,Canagliflozin,Empagliflozin,Ipragliflozin,Tofogliflozin和luseogliflozin)上市。
Scheme 1
Scheme 2
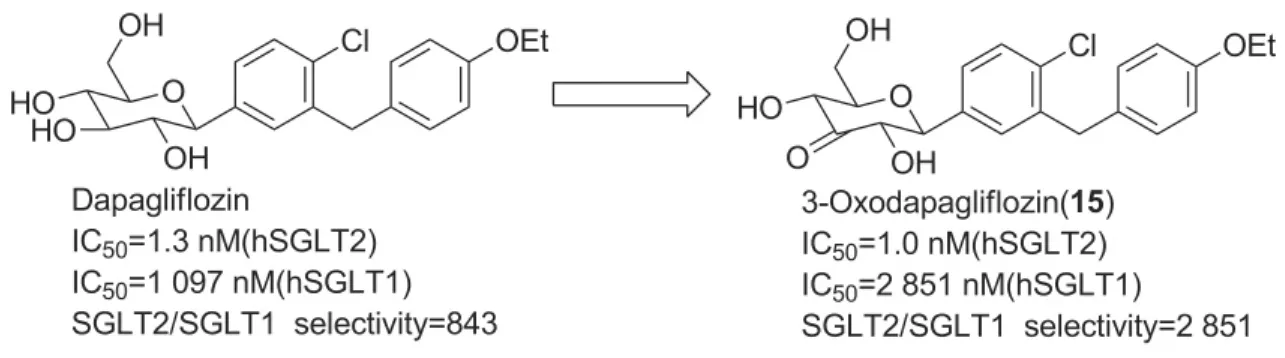
在本课题组前期的研究中,针对已经上市的达格列净(dapagliflozin)的结构衍生发现了其3-羰基衍生物,即3-羰基达格列净(15,Scheme 1),其比达格列净本身具有更强的SGLT2抑制活性和更加优异的SGLT2/SGLT1选择性,有着较好的开发前景[3]。
在15的开发研究中,有必要探索一条成本低、工艺可靠的合成路线。15在发现阶段使用的合成路线[3]以达格列净为原料,共5步反应。该合成路线步骤较少,但达格列净价格昂贵,因此需要寻找一条全新的路线。
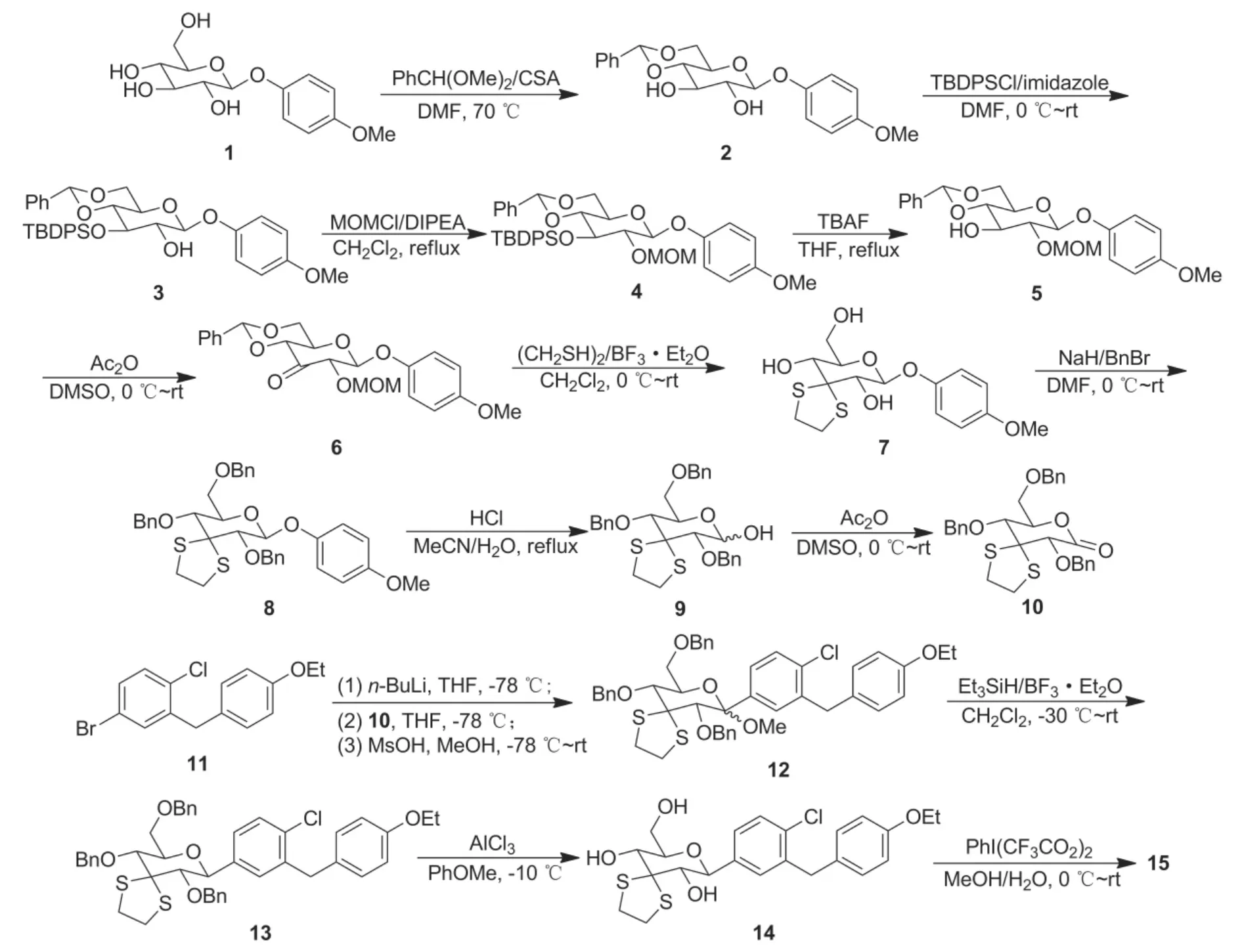
根据我们前期的研究成果[4-6],本文设计了一条汇聚式的、使用乙二硫醇保护的3-羰基葡萄糖酸内酯作为关键中间体的合成路线。以4-甲氧基苯基β-D-吡喃葡萄糖苷(1)为原料,经9步反应制得新型关键中间体2,4,6-三-O-苄基-3-羰基-D-葡萄糖酸内酯乙二硫醇缩酮(10); 10与5-溴-2-氯-4'-乙氧基二苯甲烷(11)经亲核加成反应制得甲基糖苷(12); 12在BF3·Et2O催化下经Et3SiH还原、AlCl3脱去苄基、再经双(三氟乙酰氧基)碘苯氧化脱除乙二硫醇保护基合成了15(Scheme 2),共13步反应,总收率9%。该合成路线包含了11个新化合物的合成,其结构经1H NMR,13C NMR,IR和HR-ESI-MS表征。
1 实验部分
1.1仪器与试剂
X-4型显微熔点仪(温度未校正); Bruker AV400型核磁共振仪(DMSO-d6为溶剂,TMS为内标); Bruker Vector 22型傅里叶变换红外光谱仪(KBr压片法或薄膜法); Agilent Q-TOF 6510型高分辨质谱仪(ESI)。
1[7]和11[8-9]按文献方法合成;其余所用试剂均为分析纯,溶剂用前经无水处理。
1.2合成
(1)4-甲氧基苯基4,6-O-苄叉-β-D-吡喃葡萄糖苷(2)的合成
在反应瓶中依次加入DMF 200 mL,1 28.63 g(100 mmol),PhCH(OMe)230.44 g(200 mmol)和樟脑磺酸(CSA)2.00 g,搅拌使其溶解;在N2气氛中于70℃反应12 h(TLC检测)。冷却,倾入600 mL冰水中,搅拌,迅速用CH2Cl2(3×200 mL)萃取,合并萃取液,依次用2% NaHCO3溶液(100 mL)和10%食盐水(200 mL)洗涤,无水Na2SO4干燥,旋蒸除溶后经硅胶柱层析[洗脱剂:A=V(乙酸乙酯)∶V(石油醚)=2∶1]纯化得白色固体2 30.32 g,产率81%,m.p.218℃~219 ℃;1H NMR δ:7.46~7.44(m,2H),7.40~7.36(m,3H),6.99(d,J=8.8 Hz,2H),6.86(d,J=9.2 Hz,2H),5.59(s,1H),5.55(d,J=5.6 Hz,1H),5.39(d,J=5.2 Hz,1H),4.96(d,J=7.6 Hz,1H),4.20(dd,J=4.8 Hz,10.0 Hz,1H),3.73~3.68(m,4H),3.60~3.52(m,2H),3.45(t,J=9.2 Hz,1H),3.36~3.32(m,1H)。
(2)4-甲氧基苯基-3-O-叔丁基二苯基硅基-4,6-O-苄叉-β-D-吡喃葡萄糖苷(3)的合成
在反应瓶中依次加入DMF 300 mL,2 29.95 g(80 mmol)和咪唑18.38 g(270 mmol),搅拌使其溶解;冰水浴冷却,搅拌下缓慢滴加叔丁基二苯基氯硅烷(TBDPSCl)29.68 g(108 mmol)的DMF(100 mL)溶液,滴毕,于室温反应过夜(TLC检测)。倾入1.5 L冰水中,搅拌,用二氯甲烷(3× 500 mL)萃取,合并萃取液,依次用10%食盐水(3×300 mL)洗涤,无水Na2SO4干燥,旋蒸除溶,残余物经硅胶柱层析(洗脱剂:A=1∶6)纯化得白色泡沫状固体3 41.18 g,产率84%;1H NMR δ:7.65~7.61(m,4H),7.38~7.33(m,2H),7.31~7.24(m,5H),7.17(t,J=7.4 Hz,2H),7.00(d,J=9.2 Hz,2H),6.90(d,J=7.6 Hz,2H),6.84(d,J=9.2 Hz,2H),5.67(d,J=6.0 Hz,1H),5.38(s,1H),4.93(d,J=7.6 Hz,1H),4.14(dd,J=4.8 Hz,10.0 Hz,1H),3.84(t,J=8.8 Hz,1H),3.71~3.60(m,3H),3.69(s,3H),3.48~3.42(m,1H),0.96(s,9H);13C NMR δ:154.51,151.02,137.10,135.83,135.35,134.17,132.93,129.30,129.12,128.32,127.45,127.18,127.10,125.91,117.84,114.36,101.65,100.18,80.56,75.42,74.58,67.66,65.08,55.29,26.82,19.33; IR ν:3 497,3 070,1 507,1 465,1 387 cm-1。
(3)4-甲氧基苯基-3-O-叔丁基二苯基硅基-4,6-O-苄叉-2-O-甲氧甲基-β-D-吡喃葡萄糖苷(4)的合成
在反应瓶中依次加入二氯甲烷400 mL,3 36.77 g(60 mmol),氯甲醚(MOMCl)14.49 g(180 mmol)和二异丙基乙基胺(DIPEA)46.52 g(360 mmol),搅拌使其溶解;回流反应5 h(TLC检测)。冷却,倾入500 mL冰水中,搅拌,分液,水相用二氯甲烷(2×200 mL)萃取,合并萃取液和有机相,依次用2%盐酸(2×500 mL)和10%食盐水(2× 500 mL)快速洗涤,无水Na2SO4干燥,旋蒸除溶得4粗品,直接进行下步反应。
(4)4-甲氧基苯基-4,6-O-苄叉-2-O-甲氧甲基-β-D-吡喃葡萄糖苷(5)的合成
将4粗品(按60 mmol计)溶于干燥THF(200 mL)中,搅拌下于室温加入四丁基氟化铵(TBAF)23.53 g(90 mmol)的THF(90 mL)溶液,加毕,回流反应2 h(TLC监测)。冷却,倾入800 mL冰水中,搅拌,用二氯甲烷(3×200 mL)萃取,合并萃取液,依次用10%食盐水(500 mL)洗涤,无水Na2SO4干燥,旋蒸除溶,残余物经硅胶柱层析(洗脱剂:A=1∶2)纯化,用混合溶剂(A=1∶7)重结晶得白色固体5 19.33 g,合并产率77%(3→5),m.p.159℃~160.5℃;1H NMR δ:7.46~7.44(m,2H),7.40~7.36(m,3H),6.98(d,J=8.8 Hz,2H),6.86(d,J=9.2 Hz,2H),5.60(s,1H),5.58(d,J=6.0 Hz,1H),5.12(d,J=8.0 Hz,1H),4.82(d,J=8.8 Hz,1H),4.80(d,J=8.0 Hz,1H),4.21(dd,J=4.4 Hz,9.6 Hz,1H),3.73~3.60(m,6H),3.51~3.46(m,2H),3.34(s,3H);13C NMR δ:154.54,150.67,137.62,128.82,127.98,126.29,117.29,114.55,100.67,100.44,96.28,80.28,78.28,72.25,67.73,65.51,55.33,55.12; IR ν:3 225,3 067,1 510,1 468,1 451,1 375 cm-1; HR-ESI-MS m/z:Calcd for C22H27O8{[M + H]+} 419.170 0,found 419.168 5,calcd for C22H26O8Na{[M + Na]+} 441.152 0,found 441.153 3。
(5)4-甲氧基苯基-4,6-O-苄叉-2-O-甲氧甲基-3-羰基-β-D-吡喃葡萄糖苷(6)的合成
在反应瓶中依次加入DMSO 100 mL和5 16.74 g(40 mmol),冰水浴冷却,搅拌下缓慢滴加Ac2O 50 mL,滴毕,于室温反应过夜(TLC检测)。倾入500 mL冰水中,搅拌1 h,用二氯甲烷(3× 200 mL)萃取,合并萃取液,用饱和NaHCO3溶液洗涤至水相pH>7,依次用10%食盐水(200 mL)洗涤,无水Na2SO4干燥,旋蒸除溶,残余物经硅胶柱层析(洗脱剂:A=1∶6)纯化得白色固体6 15.16 g,产率91%,m.p.160℃~161℃;1H NMR δ:7.46~7.44(m,2H),7.42~7.40(m,3H),7.03(d,J=8.8 Hz,2H),6.89(d,J=8.8 Hz,2H),5.71(s,1H),5.39(d,J=7.6 Hz,1H),4.77(d,J=14.4 Hz,1H),4.75(d,J=14.4 Hz,1H),4.68(d,J=8.8 Hz,1H),4.55(dd,J=0.8 Hz,7.4 Hz,1H),4.39(d,J=5.6 Hz,1H),3.96~3.87(m,2H),3.72(s,3H),3.36(s,3H);13C NMR δ:196.68,154.88,150.43,137.00,129.06,128.13,126.19,117.54,114.62,101.49,100.33,95.63,80.66,79.66,68.15,65.43,55.36,55.15。
(6)4-甲氧基苯基-3-羰基-β-D-吡喃葡萄糖苷乙二硫醇缩酮(7)的合成
在反应瓶中依次加入二氯甲烷100 mL,6 14.57 g(35 mmol)和乙二硫醇6.59 g(70 mmol),搅拌使其溶解;冰水浴冷却,搅拌下缓慢滴加BF3·Et2O 4.97 g(35 mmol),滴毕,于室温反应过夜(TLC监测)。搅拌下倾入200 mL冰水中,分出有机相,水相用二氯甲烷(200 mL)萃取,合并萃取液,依次用饱和NaHCO3溶液(100 mL)和10%食盐水(200 mL)洗涤,无水Na2SO4干燥,旋蒸除溶后经硅胶柱层析(洗脱剂:A=1∶2)纯化得白色泡沫固体7 9.97 g,产率79%;1H NMR δ:6.96(d,J=9.2 Hz,2H),6.84(d,J=9.2 Hz,2H),5.51(d,J=6.4 Hz,1H),5.01(d,J=6.8 Hz,1H),4.63(d,J=7.6 Hz,1H),4.55(t,J=5.6 Hz,1H),3.69(s,3H),3.67~3.65(m, 1H),3.55~3.47(m,3H),3.32~3.28(m,2H),3.27~3.21(m,4H);13C NMR δ:154.39,151.34,117.72,114.42,102.46,79.14,74.60,71.12,60.88,55.32,40.59,38.63。
(7)4-甲氧基苯基-2,4,6-三-O-苄基-3-羰基-β-D-吡喃葡萄糖苷乙二硫醇缩酮(8)的合成
在反应瓶中依次加入DMF 100 mL和7 9.01 g(25 mmol),冰水浴冷却,搅拌下缓慢分10批加入NaH 5.00 g(125 mmol),加毕,反应1 h;分3批加入BnBr 17.10 g(100 mmol),加毕,于室温反应过夜(TLC监测)。缓慢倾入500 mL冰水中,搅拌,用二氯甲烷(3×200 mL)萃取,合并萃取液,依次用10%食盐水(500 mL)洗涤,无水Na2SO4干燥,旋蒸除溶后经硅胶柱层析(洗脱剂:A=1∶4)纯化得白色固体8 14.04 g,产率89%,m.p.98.5℃~100℃;1H NMR δ:7.39~7.26(m,15H),6.99(d,J=9.2 Hz,2H),6.84(d,J=8.8 Hz,2H),4.98(d,J=10.8 Hz,1H),4.90(d,J=10.4 Hz,1H),4.84~4.81(m,2H),4.64(d,J=10.8 Hz,1H),4.56(d,J=12.0 Hz,1H),4.49(d,J=12.0 Hz,1H),3.85(d,J=8.8 Hz,1H),3.80(d,J=7.6 Hz,1H),3.72~3.67(m,3H),3.70(s,3H),3.28~3.26(m,2H),3.24~3.20(m,2H);13C NMR δ:154.60,150.99,138.42,138.11,138.07,128.21,128.19,128.13,127.90,127.79,127.63,127.60,127.48,127.42,117.48,114.55,102.50,82.12,78.96,76.79,76.76,75.60,75.40,72.34,68.88,55.33,40.44; IR ν:3 086,3 065,3 029,1 508,1 467,1 453,1 386 cm-1。
(8)2,4,6-三-O-苄基-3-羰基-β-D-吡喃葡萄糖乙二硫醇缩酮(9)的合成
在反应瓶中加入乙腈240 mL和8 12.62 g(20 mmol),搅拌使其溶解;加入6 mol·L-1盐酸24 mL,回流反应3 h(TLC检测)。倾入800 mL冰水中,搅拌,用二氯甲烷(3×200 mL)萃取,合并萃取液,依次用饱和NaHCO3溶液和10%食盐水(200 mL)洗涤,无水Na2SO4干燥,旋蒸除溶得油状物9,直接进行下步反应。
(9)10的合成
将9溶于100 mL DMSO中,冰水浴冷却,搅拌下缓慢滴加Ac2O 50 mL,滴毕,于室温反应过夜(TLC检测)。倾入500 mL冰水中,搅拌1 h,用二氯甲烷(3×200 mL)萃取,合并萃取液,用饱和NaHCO3溶液洗涤至pH>7,依次用10%食盐水(200 mL)洗涤,无水Na2SO4干燥,旋蒸除溶后经硅胶柱层析(洗脱剂:A=1∶5)纯化得无色油状物10 8.05 g,产率77%(8→10);1H NMR δ:7.43~7.28(m,15H),4.96~4.90(m,2H),4.70(d,J=11.2 Hz,1H),4.57~4.46(m,4H),4.34~4.26(m,2H),3.67(dd,J=1.8 Hz,11.4 Hz,1H),3.62(dd,J=3.4 Hz,11.4 Hz,1H),3.42~3.28(m,4H);13C NMR δ:167.72,137.78,137.75,137.59,128.28,128.21,128.14,127.94,127.77,127.75,127.65,127.61,80.55,80.43,76.51,75.22,74.39,73.45,72.31,68.44,40.17,39.75。
(10)2,4,6-三-O-苄基-1-[4-氯-3-(4-乙氧基苄基)苯基]-3-羰基-α/β-D-甲基吡喃葡萄糖苷乙二硫醇缩酮(12)的合成
在反应瓶中依次加入5-溴-2-氯-4'-乙氧基二苯甲烷(11)4.88 g(15 mmol)和THF 50 mL,氮气吹扫后用橡胶软塞封口,液氮-乙醇体系中冷却至-78℃,搅拌下缓慢滴加正丁基锂9.4 mL(15 mmol),滴毕,反应1 h;缓慢滴加10 7.84 g(15 mmol)的THF(40 mL)溶液,滴毕,反应1 h;缓慢滴加甲磺酸4.81 g(50 mmol)的甲醇(20 mL)溶液,滴毕,于室温反应过夜。倾入400 mL冰水中,搅拌,用二氯甲烷(3×100 mL)萃取,合并萃取液,依次用饱和NaHCO3溶液(100 mL)和10%食盐水(100 mL)洗涤,无水Na2SO4干燥,旋蒸除溶得12粗品,不经纯化,直接进行下步反应。
(11)2,4,6-三-O-苄基-1-[4-氯-3-(4-乙氧基
苄基)苯基]-1-脱氧-3-羰基-β-D-吡喃葡萄糖乙二硫醇缩酮(13)的合成
在反应瓶中依次加入二氯甲烷100 mL和12粗品(按15 mmol计),搅拌使其溶解;加入Et3SiH 3.49 g(30 mmol),氮气气氛中冷却至-30℃,搅拌下缓慢滴加BF3·Et2O 2.13 g(15 mmol)的二氯甲烷(5 mL)溶液,滴毕,反应30 min;于室温反应过夜(TLC检测)。搅拌下缓慢加入饱和NaHCO3溶液100 mL,搅拌30 min,分出有机相,水相用二氯甲烷(2×100 mL)萃取,合并有机相和萃取液,依次用10%食盐水(200 mL)洗涤,无水Na2SO4干燥,旋蒸除溶后经硅胶柱层析(洗脱剂:A=1∶10)纯化得无色油状液体13 7.80 g,产率69%(10→13);1H NMR δ:7.44(d,J=8.0 Hz,1H),7.39(d,J=1.6 Hz,1H),7.35~7.28(m, 11H),7.22~7.20(m,3H),7.04(d,J=8.4 Hz,2H),6.90~6.88(m,2H),6.71(d,J=8.4 Hz,2H),4.92(d,J=10.8 Hz,1H),4.68(d,J=10.4 Hz,1H),4.59~4.47(m,3H),4.16(d,J=9.2 Hz,1H),4.02~3.88(m,5H),3.79(d,J=9.2 Hz,1H),3.71(dd,J=4.0 Hz,11.2 Hz,1H),3.66~3.64(m,2H),3.53~3.50(m,1H),3.27~3.24(m,2H),3.20~3.19(m,2H),1.26(t,J=7.0 Hz,3H);13C NMR δ:156.83,138.50,138.40,138.24,138.18,137.78,132.56,131.05,130.90,129.45,129.10,128.21,127.99,127.89,127.65,127.57,127.51,127.42,127.35,127.29,114.22,84.25,81.28,79.87,79.25,78.47,75.44,75.19,72.49,69.17,62.81,39.98,38.98,37.51,14.61。
(12)1-[4-氯-3-(4-乙氧基苄基)苯基]-1-脱氧-3-羰基-β-D-吡喃葡萄糖乙二硫醇缩酮(14)的合成
在反应瓶中依次加入苯甲醚75 mL和13 7.53 g(10 mmol),搅拌使其溶解;冷却至-10℃,分10批加入无水AlCl36.67 g(50 mmol),加毕,反应30 min;于室温反应2 h(TLC检测)。搅拌下缓慢倾入300 mL冰水中,用二氯甲烷(3× 100 mL)萃取,合并萃取液,依次用2%盐酸(200 mL)和10%食盐水(2×200 mL)洗涤,无水Na2SO4干燥,旋蒸除去二氯甲烷,油泵减压蒸除苯甲醚得14粗品,经硅胶柱层析(洗脱剂:A=1∶2)纯化得白色泡沫状固体14 5.88 g,产率78%;1H NMR δ:7.36~7.34(m,2H),7.25(dd,J=2.0 Hz,8.0 Hz,1H),7.09(d,J=8.8 Hz,2H),6.81(d,J=8.4 Hz,2H),4.96(d,J=6.8 Hz,1H),4.91(d,J=7.2 Hz,1H),4.43(t,J=6.0 Hz,1H),4.01~3.93(m,5H),3.70~3.66(m,1H),3.48(dd,J=6.8 Hz,9.6 Hz,1H),3.50~3.46(m,2H),3.29~3.20(m,5H),1.28(t,J=7.0 Hz,3H);13C NMR δ:156.84,139.25,137.72,132.03,131.17,131.10,129.51,128.64,127.34,114.23,81.84,81.74,80.84,76.14,71.58,62.83,61.38,40.21,38.61,37.62,14.63; IR ν:3 445,3 031,1 611,1 582,1 510,1 477,1 392 cm-1。
(13)15的合成
在反应瓶中依次加入甲醇21 mL,水3 mL和14 2.42 g(5 mmol),搅拌使其溶解;冰水浴冷却,搅拌下分5批加入双(三氟乙酰氧基)碘苯8.60 g(20 mmol),加毕,于室温反应5 h(TLC检测)。搅拌下倾入100 mL冰水中,用二氯甲烷(3×50 mL)萃取,合并萃取液,依次用10%食盐水洗涤,无水Na2SO4干燥,旋蒸除溶,残余物经硅胶柱层析(洗脱剂:A=1∶2)纯化得白色泡沫状固体15 1.36 g,产率67%;1H NMR δ:7.43~7.41(m,2H),7.33(dd,J=2.2 Hz,8.2 Hz,1H),7.08(d,J=8.4 Hz,2H),6.82(d,J=8.4 Hz,2H),5.38(d,J=6.0 Hz,1H),5.31(d,J=6.0 Hz,1H),4.75(t,J=6.0 Hz,1H),4.24~4.12(m,3H),3.99~3.93(m,4H),3.73(dd,J=5.2 Hz,10.4 Hz,1H),3.61~3.57(m,1H),3.40(dd,J=3.2 Hz,10.0 Hz,1H),1.28(t,J=7.0 Hz,3H)。与文献[3]报道一致。
2 结果与讨论
2.1合成
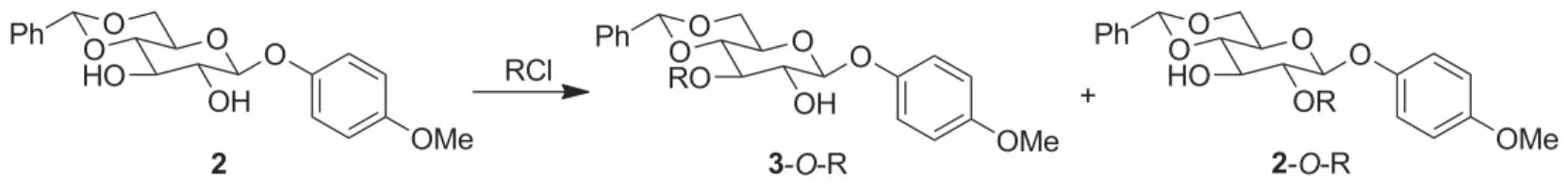
起始原料1按照文献[7]方法使用D-葡萄糖和4-甲氧基苯酚经多步反应制得。1与苯甲醛二甲基缩醛在CSA的催化下在DMF中于70℃反应,其糖环上的4-OH和6-OH被苄叉同时保护制得二羟基化合物2。2与1.35 eq.TBDPSCl在咪唑作用下在DMF中于室温反应,其糖环上的3-OH被区域选择性地保护制得2-OH游离的化合物3。对于2中糖环上的3-OH在2-OH存在下的选择性保护(Scheme 3),我们系统地筛选和研究了常见的含硅的保护基,结果见表1。由表1可见,随着保护基体积增大(TBDMS→TIPS→TBDPS),2中糖环上3-OH被保护所得产物(3-OR)的比例越来越高,因此本文最终选择TBDPS。上述结果同时表明,大体积的保护基倾向于保护2中的3-OH。说明2-OH附近的4-甲氧基苯基的体积效应远大于3-OH周围的4,6-O-苄叉基,该现象与我们以前观察到的类似现象一致[10]。应该指出的是,在实际操作中3中所包含的少量2-OH被保护的异构体(2-O-TBDPS)很难被柱层析彻底除去,研究表明可以直接将粗品3用于下一步反应,相关的杂质可以在纯化5时通过柱层析和结晶除去。
对于2中糖环上3-OH被不同保护基保护所得各个主要化合物(3,3-1和3-2),本文利用NOE技术来证明该保护基团确实在3-OH上(Chart 1)。 TBDMS,TIPS和TBDPS三个保护基上的CH3上的质子与4,6-O-位上的苄叉基中苯环的邻位质子均具有NOE效应,说明它们的空间距离相近,进一步说明这三个保护基均在糖环的3-OH上;因为如果保护基在2-OH上,则会因为空间距离较远,难以观察到这种效应[10]。因此,2在使用上述三个大体积的含硅保护基保护时,主要产物中的保护基连接在糖环的3-OH上。
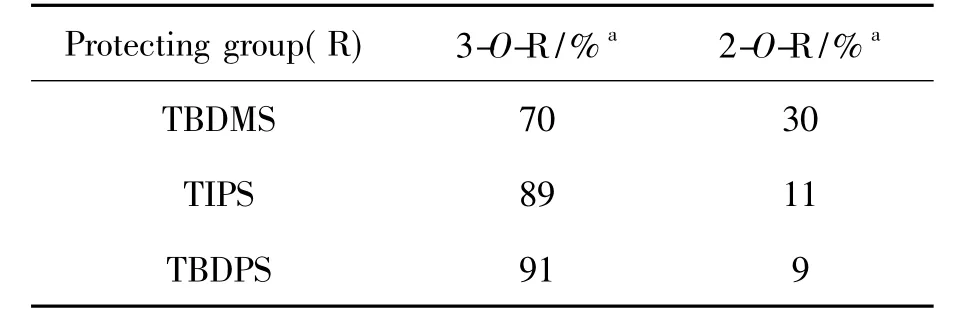
表1 不同保护基对2中2-OH和3-OH选择性保护的研究结果Table 1 The results of the exploration of protecting groups for selective protection of 2-OH and 3-OH in 2
以6 eq.DIPEA为碱,3与3 eq.MOMCl在二氯甲烷中回流反应,其2-OH被进一步保护制得4。研究表明,MOMCl用量低于3 eq.时,反应进行较慢,有时甚至难以进行。可能与3中2-OH的周围有两个体积较大基团的位阻有关。如上所述,由于用于本步反应的原料粗品3中含有少量的2-OH被TBDPS保护的异构体(即2-O-TBDPS),导致生成的4中也含有该异构体引入的相应的杂质(即2-O-TBDPS-3-O-MOM),该杂质与其异构体4很难经柱层析彻底分离,因此我们直接将粗品4用于下步反应。4与1.5 eq.TBAF在THF中回流反应顺利脱去3-OH上的TBDPS保护基得到5。5经柱层析和结晶提纯后,可彻底除去由于直接使用含少量异构体(2-O-TBDPS)的粗品3而在本步反应引入的杂质(3-O-MOM)。5于室温经Swern氧化(Ac2O/DMSO)得3-羰基化合物6,从而实现了糖环上3-位上羰基的构建。6与2 eq.乙二硫醇在BF3·Et2O催化下在二氯甲烷中于室温反应制得7,此时6中的苄叉保护基和MOM保护基也在酸性BF3·Et2O存在下均被同时脱除,由于脱除的苄叉是以乙二硫醇缩醛的形式存在,所以该反应的乙二硫醇需要2 eq.。7在DMF中经NaH/Bn-Br处理制得三个羟基均被保护的化合物8。
Scheme 3

Chart 1

Scheme 4
在最初的试验中我们也尝试了使用NaH/BnBr处理3以便得到16,并期望通过16来合成化合物8,但是该路线没有成功。因为3被NaH/BnBr使用常规方法处理后最终分离出来的产物被证明为17而非期望的产物16(Scheme 4)。该实验结果说明在强碱NaH的存在下,3中OH上的TBDPS保护基很不稳定,会被NaH脱除而重新变回OH(并接着被BnBr苄基化)。
8在乙腈/水中在盐酸催化下回流水解脱去4-甲氧基苯酚制得9,9为α/β异构体的混合物,不需提纯,直接于室温经Swern氧化(Ac2O/DMSO)顺利制得关键中间体10,从而最终实现了糖环上3-羰基的构建并使用乙二硫醇进行保护。
11按文献[8-9]方法制备。11在干燥的THF中低温下(-78℃)用1 eq.n-BuLi处理得相应的芳基锂,后者与随后加入的10经亲核加成反应制得含OH的产物,再与甲磺酸和甲醇反应制得甲基糖苷12。12是α/β的混合物,无需进一步提纯,直接在低温(-30℃~室温)在BF3·Et2O催化下经Et3SiH还原得13。13在苯甲醚于-10℃经AlCl3脱去全部苄基制得14。14在甲醇/水中用4 eq.PhI(CF3CO2)2处理,氧化脱去乙二硫醇保护基合成15,其1H NMR分析与我们前期的研究结果[3]一致,说明本文最新开发的15的合成路线可行。
3 结论
利用4-甲氧基苯基-β-D-吡喃葡萄糖苷为原料,经过关键中间体2,4,6-三-O-苄基-3-羰基-D-葡萄糖酸内酯乙二硫醇缩酮(10)制备了新型SGLT2抑制剂3-羰基达格列净,共13步反应,总收率9%。该合成路线中中间体3~10和12~14均为新化合物。关键中间体10是一个具有新颖结构的糖酸内脂,其设计和合成均为首次报道,10可作为一个通用的糖基供体类合成砌块用于芳基C-糖苷3-羰基衍生物的合成。
该合成路线具有成本低、操作简便的优点,代表了芳基C-糖苷3-羰基衍生物的一种全新的合成方法,具有成本低、操作简便的优点。
参考文献
[1]Washburn W N.Development of the renal glucose reabsorption inhibitors:A new mechanism for the pharmacotherapy of diabetes mellitus type 2[J].J Med Chem,2009,52(7):1785-1794.
[2]Diamant M,Morsink L M.SGLT2 inhibitors for diabetes:Turning symptoms into therapy[J].Lancet,2013,382(9896):917-918.
[3]Zhang S,Wang Y L,Liu W,et al.3-Oxodapagliflozin as a potent and highly selective SGLT2 inhibitor for the treatment of type 2 diabetes[J].Chem Res Chin Univ,2014,30(5):785-793.
[4]Shi Y H,Xu H Q,Liu B N,et al.A facile synthesis of 6-deoxydapagliflozin[J].Monatsh Chem,2013,144(12):1903-1910.
[5]高志刚,张大同,魏鹏,等.SGLT2抑制剂3-脱氧达格列净简便的汇聚式合成方法[J].有机化学,2014,34(9):1829-1839.
[6]韩书文,王玉丽,刘巍,等.新型Na+依赖性葡萄糖共转运体2(SGLT2)抑制剂6-脱氧canagliflozin的简便合成方法及其晶型研究[J].中国药学杂志,2014,49(20):1854-1859.
[7]Zhang Z Y,Magnusson G.Conversion of p-methoxyphenyl glycosides into the corresponding glycosyl chlorides and bromides,and into thiophenyl glycosides[J].Carbohydr Res,1996,295:41-55.
[8]Meng M,Ellsworth B A,Nirschl A A,et al.Discovery of dapagliflozin:A potent,selective renal sodium-dependent glucose cotransporter 2(SGLT2)inhibitor for the treatment of type 2 diabetes[J].J Med Chem,2008,51(5):1145-1149.
[9]邵华,赵桂龙,刘巍,等.SGLT2抑制剂Dapagliflozin的全合成[J].合成化学,2010,18(3):389-392.
[10]Zhang L Y,Wang,Y L,Xu H Q,et al.Discovery of 6-deoxydapagliflozin as a highly potent sodium-dependent glucose cotransporter 2(SGLT2)inhibitor for the treatment of type 2 diabetes[J].Med Chem,2014,10(3):
New Synthetic Route of Novel SGLT2 Inhibitor——3-Oxodapagliflozin
JIANG Lin-lin1,YIN Ling1,XU Wei-ren2,
TANG Li-da2,WANG Wen-jin2,ZHAO Gui-long2
(1.Department of Chemistry and Chemical Engineering,Jining University,Qufu 273155,China; 2.Tianjin Key Laboratory of Molecular Design and Drug Discovery,Tianjin Institute of Pharmaceutical Research,Tianjin 300193,China)
Abstract:The key intermediate——2,4,6-tri-O-benzyl-3-oxo-D-gluconolactone ethylene dithioketal(10),was prepared by nine-step reaction from 4-methoxyphenyl β-D-glucopyranoside.Methyl 2,4,6-tri-O-benzyl-1-[4-chloro-3-(4-ethoxybenzyl)phenyl]-3-oxo-α/β-D-glucopyranoside ethylene dithioketal(12)was synthesized by nucleophilic addition of 10 with 5-bromo-2-chloro-4'-ethoxybiphenyl.The novel SGLT2 inhibitor,3-oxodapagliflozin,was synthesized by reduction and deprotection from 12.The overall yield was 9%.Eleven novel compounds were involved in the synthetic route.The structures were characterized by1H NMR,13C NMR,IR and HR-ESI-MS.
Keywords:SGLT2 inhibitor; dapagliflozin; 3-oxodapagliflozin; synthetic methodology
作者简介:姜琳琳(1980-),女,汉族,辽宁海城人,硕士,讲师,主要从事药物化学研究。
基金项目:国家自然科学基金资助项目(21302141);山东省自然科学基金资助项目(ZR2015BM028);天津市应用基础与前沿
收稿日期:2014-12-23;
修订日期:2015-08-27
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.11.1005 *
文献标识码:A
中图分类号:O621.3; O629.13
