非稳定型亚甲胺基叶立德参与的1,3-偶极环加成反应合成新型5-取代噁唑烷衍生物*
2016-01-17曾凡浩徐亮四川大学华西药学院四川成都610041
曾凡浩,徐亮(四川大学华西药学院,四川成都 610041)
非稳定型亚甲胺基叶立德参与的
1,3-偶极环加成反应合成新型5-取代噁唑烷衍生物*
曾凡浩,徐亮
(四川大学华西药学院,四川成都610041)
摘要:以N-三甲基硅甲基苄胺,多聚甲醛和芳香醛为原料,磷酸为催化剂,经1,3-偶极环加成反应合成了6个5-取代噁唑烷衍生物(3a~3f,其中3c~3f为新化合物),收率57%~73%,转化率70%~90%,其结构经1H NMR,13C NMR和ESI-MS表征。
关键词:N-三甲基硅甲基苄胺; 1,3-偶极环加成反应;合成;噁唑烷衍生物
亚甲胺基叶立德参与的1,3-偶极环加成反应是构建五元含氮杂环的重要方法之一,该方法已广泛应用于相关天然产物的合成中[1-4]。虽然稳定型亚甲胺基叶立德参与的三组分1,3-偶极环加成反应的研究已取得诸多成果[5-11],但非稳定型亚甲胺基叶立德参与的三组分1,3-偶极环加成反应却鲜有报道[12]。Risch课题组[13]利用二苄胺与苯甲醛在甲苯中回流,再与多种亲偶极体发生1,3-偶极环加成反应,合成了一系列多芳基取代的五元含氮衍生物。Torii[14]将N-三甲基硅甲基苄胺(1)与醛在THF中回流反应,再与贫电子烯烃发生三组分1,3-偶极环加成反应制得四氢吡咯烷衍生物。上述生成非稳定亚甲胺基叶立德的方法所需反应条件均较苛刻。本研究组曾报道了一种以1,多聚甲醛和取代烯烃为原料,经三组分1,3-偶极环加成反应,于温和条件下高收率合成一系列3,4-二取代四氢吡咯衍生物的方法[15]。
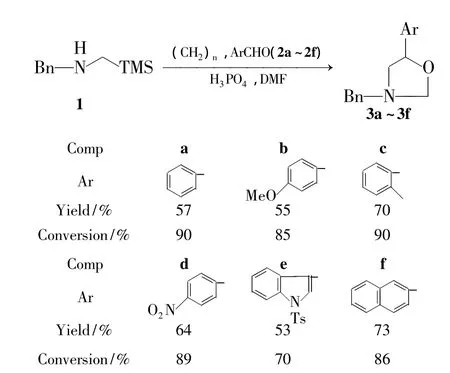
Scheme 1
在此基础上,本文以1,多聚甲醛和芳香醛(2b~2f)为原料,磷酸为催化剂,经1,3-偶极环加成反应,合成了6个5-取代噁唑烷衍生物(3a~3f,其中3c~3f为新化合物,Scheme 1),收率57%~73%,转化率70%~90%,其结构经1H NMR,13C NMR和ESI-MS表征。
1 实验部分
1.1仪器与试剂
Varian Unity NOVA-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标); Waters Q-TOFPremier型质谱仪。
1按文献[16]方法合成[收率91%;1H NMR δ:8.15(s,1H),7.72~7.38(m,5H),3.42(s,2H),0.08(s,9H)];其余所用试剂均为分析纯。
1.2 3a~3f合成(以3a为例)
氩气保护,在干燥的反应管中依次加入1 290 mg(1.5 mmol),多聚甲醛45 mg(1.5 mmol)和无水DMF 2 mL,搅拌使其溶解;加入苯甲醛(2a)106mg(1 mmol)和磷酸6 μL(0.1 mmol),于室温反应48 h。加入乙酸乙酯2 mL,依次用饱和NaHCO3溶液(5 mL)和饱和NaCl溶液(5 mL)洗涤,用无水Na2SO4干燥,蒸除溶剂后经硅胶柱层析[洗脱剂:V(石油醚)∶V(乙酸乙酯)=4∶1]纯化得无色液体3-苄基-5-苯基噁唑烷(3a)[17];1H NMR δ:7.37~7.26(m,10H),5.06(t,J=7.2 Hz,1H),4.61~4.59(m,2H),3.83(s,2H),3.43(dd,J=6.8 Hz,11.6 Hz,1H),2.83(dd,J=8.0 Hz,11.6 Hz,1H); ESI-MS m/z:240.13{[M + H]}+。
分别以2b~2f替代2a,用类似的方法合成无色液体3b~3f。
3-苄基-5-对甲氧基苯基噁唑烷(3b)[17]:
1H NMR δ:7.36~6.87(m,9H),5.01(t,J=7.2 Hz,1H),4.59~4.56(m,2H),3.82(s,2H),3.80(s,3H),3.38(dd,J=6.8 Hz,11.6 Hz,1H),2.81(dd,J=8.0 Hz,11.6 Hz,1H); ESI-MS m/z:270.14{[M + H]+}。
3-苄基-5-邻甲苯基噁唑烷(3c):1H NMR δ:7.55~7.11(m,9H),5.23(t,J=7.2 Hz,1H),3.84(s,2H),3.50(dd,J=6.8 Hz,11.2 Hz,1H),2.70(dd,J=7.6 Hz,11.2 Hz,1H),2.26(s,3H);13C NMR δ:140.5,138.7,134.0,130.2,128.7,128.7,128.4,128.4,127.3,127.0,126.2,124.3,87.1,74.1,59.3,58.5,19.2; ESI-MS m/z:254.15{[M + H]+}。
3-苄基-5-对硝基苯基噁唑烷(3d):1H NMR δ:8.20(d,J=8.8 Hz,2H),7.49(d,J=8.4 Hz,2H),7.34~7.25(m,5H),5.12(t,J=7.2 Hz,1H),4.61(s,2H),3.80(s,2H),3.47(dd,J=6.8 Hz,11.6 Hz,1H),2.78(dd,J=7.2 Hz,11.6 Hz,1H);13C NMR δ:150.0,138.2,128.8,128.6,128.6,128.5,128.5,127.5,126.1,126.1,123.8,123.8,87.8,75.5,60.3,58.2; ESI-MS m/z:285.12{[M +H]+}。
3-苄基-5-(N-对甲苯磺酰基-1H-吲哚)-噁唑烷(3e):1H NMR δ:8.00~7.17(m,14H),5.26~5.23(m,1H),4.59~4.56(m,2H),3.84(s,2H),3.43(dd,J=6.8 Hz,11.2 Hz,1H),3.00(dd,J=7.6 Hz,11.2 Hz,1H),2.33(s,3H);13C NMR δ:144.9,138.5,135.5,135.0,129.8,129.8,129.7,128.6,128.6,128.4,128.4,127.3,126.7,126.7,124.8,123.3,123.2,122.5,120.0,113.7,86.8,70.5,58.3,57.9,21.5; ESI-MS m/z:433.15{[M +H]+}。
3-苄基-5-(2-萘基)-噁唑烷(3f):1H NMR δ:
7.89~7.26(m,12H),5.76(t,J=7.2 Hz,1H),4.71~4.67(m,2H),3.94~3.85(m,2H),3.74(dd,J=7.2 Hz,11.6 Hz,1H),2.88(dd,J=7.6 Hz,11.6 Hz,1H);13C NMR δ:138.7,138.1,133.7,130.1,128.9,128.7,128.7,128.4,128.4,127.5,127.3,126.0,125.7,125.5,122.9,121.4,87.0,74.0,60.0,58.6; ESI-MS m/z:290.15{[M + H]+}。
2 结果与讨论
2.1芳醛底物的拓展
以10 mol% H3PO4为催化剂,DMF为溶剂,其余反应条件同1.2(2),考察了醛类拓展底物(2b~2f)的适应性,结果见Scheme 1。由Scheme 1可见,无论是富电子或贫电子取代基的苯甲醛(2b,2c和2f),还是稠环芳烃与芳杂环取代的醛(2d和2e),均能以较高收率合成最终产物。说明该反应条件对取代芳醛有良好的底物适应性。
2.1构建噁唑烷的新方法
Padwa等[18]报道了非稳定型亚甲胺基叶立德与醛在氟化锂作用下进行二组分1,3-偶极环加成反应的方法。本文方法与二组分方法相比,省略了不易制备和贮存的亚甲胺基叶立德前体的合成步骤,更为直接和高效。
参考文献
[1]Paul R S,Hiroyuki O,Robert M W.Asymmetric,stereocontrolled total synthesis of(+)and(-)-spirotryprostatin B via a diastereoselective azomethine ylide [1,3]-dipolar cycloaddition reaction[J].Tetrahedron,2002,58:6311-6322.
[2]Tony K,Yong L Z.Ring-selective synthesis of O-heterocycles from acyclic 3-O-allyl-monosaccharides via intramolecular nitrone-alkene cycloaddition[J].Tetrahedron,2001,57:1573-1579.
[3]James M,Bin W.Highly enantioselective Ag(I)-catalyzed[3 +2]cycloaddition of azo-methine ylides[J].J Am Chem Soc,2002,124:13400-13401.
[4]Ian S Y,Michael A K.Three-component homo 3 + 2 dipolar cycloaddition:A diversity-oriented synthesis of tetrahydro-1,2-oxazines and FR900482 skeletal congeners[J].Organic Letters,2004,6:139-141.
[5]Williams R M,Zhai W,Aldous D J,et al.Asymmetric[1,3]-dipolar cyclo-addition reaction:Synthesis of highly substituted proline derivatives[J].J Org Chem,1992,57:6527-6532.
[6]Hamper B C,Dukesherer D R,South M S.Solidphase synthesis of proline analogs via a three component 1,3-dipolar cycloa-ddition[J].Tetrahedron Letters,1996,37:3671-3674.
[7]Sebahar P R,Williams R M.The asymmetric total synthesis of(+)- and(-)-spiroo-tryprostatin B [J].J Am Chem Soc,2000,122:5666-5667.
[8]Ahrendt K A,Williams R M.A concise asymmetric synthesis of the ADE fragment of nakadomarin A[J].Organic Letters,2004,6:4539-4541.
[9]Wilson N S,Sarko C R,Roth G P.Microwave-assisted synthesis of a[3 + 2]cyclo-addition library[J].Tetrahedron Lett,2001,42:8939-8941.
[10]Li G Y,Chen J,Yu W Y,et al.Stereo-selective synthesis of functionalized pyrrolidines by ruthenium porphyrin-catalyzed decomposition of α-diazo esters and cascade azomethine ylide formation 1,3-dipolar cycloaddition reactions[J].Organic Letters,2003,5:2153-2156.
[11]Galliford C V,Beenen M A,Nguyen S T,et al.Catalytic,three-cimponent assembly reaction for the synthesis of pyrrolidines[J].Organic Letters,2003,5:3487-3490.
[12]Pearson W H,Stoy P,Mi Y.A three-component,one-pot synthesis of indolizidines and related heterocycles via the[3 + 2]cycloadditon of nonstabilized azomethine ylides[J].J Org Chem,2004,69:1919-1939.
[13]Wittland C,Arend M,Risch N.Synthesis of 2,4,5-triphenyl-substituted oxazolidine and 2,5-diphenylsubstituted pyrrolidine[J].Synthsis,1996,3:367-371.
[14]Torii S,Okumoto H,Genba A.A versatile cycloaddition for the generation of pyrroli-dine derivatives via C-N-C 1,3-dipoles[J].Chemistry Letters,1996,9:747-748.
[15]潘华,徐亮.非稳定型亚甲胺基叶立德参与的三组分1,3-偶极环加成反应合成新型四氢吡咯衍生物[J].合成化学,2012,20(4):466-469.
[16]Albert P,William D.N-benzyl-N-methoxymethyl-N-(trimethylsilyl)methylamine as an azomethine ylide equivalent:2,6-Dioxo-1-phenyl-4-benzyl-1,4-diazabicyclo[3.3.0]octane[J].Org Synth,1993,8:231-232.
[17]Ryan J H.Synthesis of 5-aryloxazolidines via 1,3-dipolar cycloaddition reaction of a non-stabilized azomethine ylide with aromatic aldehydes[J].Australian Journal of Chemistry,2007,60:898-904.
[18]Albert P,William D.On the use of N-(trimethylsilylmethyl)amine ethers as capped azomethine ylide equivalents[J].J Org Chem,1987,52:235-244.
·研究简报·
Syntheses of Novel Oxazolidine Derivatives by 1,3-Dipolar Cycloaddition Reaction by Unstabilized Azomethine Ylides
ZENG Fan-hao,XU Liang
(West China School of Pharmacy,Sichuan University,Chengdu 610041,China)
Abstract:Six oxazolidine derivatives(3a~3f,3c~3f were novel compounds),in yield of 57%~73% and conversion of 70%~90%,were synthesized by 1,3-dipolar cycloaddition reaction from N-(trimethylsilylmethyl)benzylamine,paraformaldehyde and aromatic aldehyde,using phosphoric acid as catalyst.The structures were characterized by1H NMR,13C NMR and ESI-MS.
Keywords:N-(trimethylsilylmethyl)benzylamine; 1,3-dipolar cycloaddition reaction; synthesis; oxazolidine derivative
通讯作者:徐亮,副教授,Tel.028-85503046,E-mail:liangxu@ scu.edu.cn
作者简介:曾凡浩(1990-),男,汉族,江西赣州人,硕士研究生,主要从事天然药物合成的研究。E-mail:zfanhao@163.com
基金项目:国家自然科学基金资助项目(21472129)
收稿日期:2015-03-04
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.06.0522 *
文献标识码:A
中图分类号:O626.13; O626.24
