新型邻菲啰啉衍生物的合成*
2016-01-17张灯青李贤英向芸颉金武松东华大学a化学化工与生物工程学院环境科学与工程学院上海201620
李 荣,王 强,张灯青,李贤英,向芸颉,金武松(1.东华大学a.化学化工与生物工程学院; b.环境科学与工程学院,上海 201620)
新型邻菲啰啉衍生物的合成*
李荣1a,王强1a,张灯青1a,李贤英1b,向芸颉1a,金武松1a
(1.东华大学a.化学化工与生物工程学院; b.环境科学与工程学院,上海201620)
摘要:以2-[2-(2-甲氧基乙氧基)乙氧基]乙基-4-甲基苯磺酸为原料,经2步反应制得中间体2,2'-【2,5-二{ 2-[2-(2-甲氧基乙氧基)乙氧基]}-1,4-二(4,4,5,5-四甲基)-1,3,2-二氧硼基】苯(6); 3-溴-1,10-邻菲啰啉和6 经Suzuki偶联反应合成了一个新型的邻菲啰啉衍生物——3,3'-【2,5-二{ 2-[2-(2-甲氧基乙氧基)乙氧基]}-1,4-二(1,10-菲啰啉基)】苯,其结构经1H NMR和MALDI-TOF-MS表征。
关键词:1,10-邻菲啰啉; Suzuki偶联反应;合成
邻菲啰啉及其衍生物广泛应用于光度分析[1-2]、超分子组装分子骨架[3]和光电材料[4]等领域。其中1,10-邻菲啰啉易于与金属离子形成稳定配合物,该类配合物具有良好的理化性质,常用于制备有机电致发光材料[5-7]和开发新型钌染料敏化剂[8]。
合成邻菲啰啉衍生物的主要方法有:将1,10-邻菲啰啉氧化为1,10-邻菲啰啉-5,6-二酮,再将其转化为其他衍生物[9]。该反应需用浓硝酸或浓硫酸,危险性较大。为优化邻菲啰啉衍生物的合成方法,本文以1,10-邻菲啰啉为原料,经溴代反应制得3-溴-1,10-邻菲啰啉(1);以2-[2-(2-甲氧基乙氧基)乙氧基]乙基-4-甲基苯磺酸(3)为原料,经2步反应制得中间体2,2'-【2,5-二{ 2-[2-(2-甲氧基乙氧基)乙氧基]}-1,4-二(4,4,5,5-四甲基)-1,3,2-二氧硼基】苯(6); 1和6经Suzuki偶联反应合成了一个新型的邻菲啰啉衍生物——3,3'-【2,5-二{ 2-[2-(2-甲氧基乙氧基)乙氧基]}-1,4-二(1,10-菲啰啉基)】苯(7,Scheme 1),其结构经1H NMR和MALDI-TOF-MS表征。该方法操作简单,并通过引入三甘醇单甲醚(OTEG)链有效改善了1,10-邻菲啰啉衍生物的溶解性[10-11]。
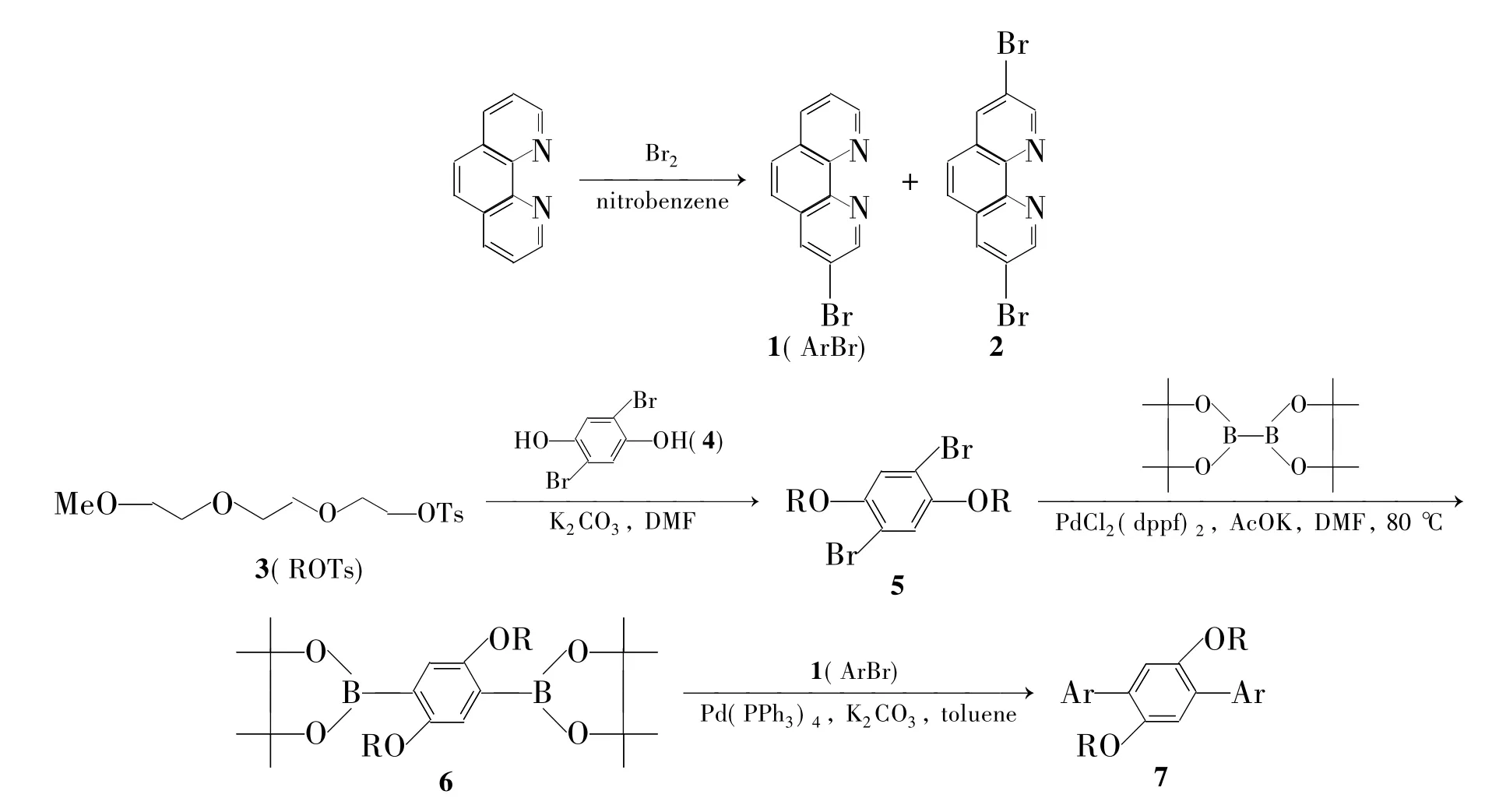
Scheme 1
1 实验部分
1.1仪器与试剂
Bruker AV-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标); ABS ciex-4800型质谱仪。
3按文献[12]方法合成;其余所用试剂均为分析纯。
1.2合成
(1)1和2的合成
在两口瓶中加入一水合1,10-邻菲啰啉4 g(20 mmol),氩气保护,搅拌下于135℃滴加硝基苯8 mL,滴毕,搅拌使其溶解;缓慢滴加Br21 mL(39 mol)的硝基苯(4 mL)溶液,滴毕,反应48 h。冷却至室温,用氨水中和后用CH2Cl2萃取,无水硫酸镁干燥。旋蒸除溶后经硅胶柱层析[洗脱剂:A=V(二氯甲烷)∶V(乙酸乙酯)=1∶10]纯化得白色固体1和2。
1:产率46.5%;1H NMR δ:9.27(dd,J=4.4 Hz,1.5 Hz,1H),9.21(d,J=2.2 Hz,1H),8.43(d,J=2.3 Hz,1H),8.34(dd,J=8.1 Hz,1.5 Hz,1H),7.87(d,J=8.9 Hz, 1H),7.78~7.71(m,2H)。
2:产率26.6%;1H NMR δ:9.18(d,J=2.1 Hz,2H),8.38(t,J=17.5 Hz,2H),7.76(s,2H)。
(2)1,4-二溴-2,5-二{ 2-[2-(2-甲氧基乙氧基)乙氧基]乙基}苯(5)的合成
在两口瓶中依次加入2,5-二溴对苯二酚(4)
1 g(3.77 mmol),3 2.4 g(7.6 mmol)和K2CO35 g(37.7 mmol),氩气保护,搅拌下加入干燥DMF 30 mL,于80℃反应10 h。冷却至室温,旋蒸除溶,剩余物用CH2Cl2萃取,无水硫酸镁干燥。旋蒸除溶后经硅胶柱层析(洗脱剂:A=1∶10)纯化得淡黄色液体5,产率85.7%;1H NMR δ:7.13(s,2H),4.10(d,J=5.1 Hz,4H),3.87~3.83(m,4H),3.76(dd,J=5.7 Hz,3.7 Hz,4H),3.68~3.63(m,9H),3.54(dd,J=5.7 Hz,3.6 Hz,4H),3.36(s,6H); MALDI-TOF-MS m/z:Calcd for C20H32O8Br2{[M + Na]+} 583.034 1,found 583.094 3。
(3)6的合成
在两口瓶中依次加入5 300 mg(0.54 mmol),双联频哪醇硼酸酯302 mg(1.19 mmol),PdCl2(dppf)240 mg(0.053 mmol)和KAc 260 mg(2.7 mmol),氩气保护,搅拌下加入干燥DMF 25 mL,于80℃反应4 h。冷却至室温,旋蒸除溶,剩余物用CH2Cl2萃取,无水硫酸镁干燥。旋蒸除溶后经硅胶柱层析(洗脱剂B:乙酸乙酯)纯化和GPC分离得黄色液体6,产率34.0%;1H NMR δ:7.13(s,2H),4.11(t,J=5.1 Hz,4H),3.85(t,J=5.0 Hz,4H),3.79(d,J=5.2 Hz,4H),3.67~3.63(m,8H),3.54(d,J=4.3 Hz,4H),3.37(s,6H),1.33(s,24H); MALDI-TOF-MS m/z:Calcd for C32H56O12B2{[M + Na]+} 677.385 6,found 677.007 2。
(4)7的合成
在两口瓶中依次加入6 100 mg(0.13 mmol),1 135 mg(0.52 mmol),Pd(PPh3)423 mg(0.02 mmol)和2 mol·L-1K2CO30.8 mL,氩气保护,搅拌下加入冷冻除氧的甲苯15 mL,于110℃反应36 h。旋蒸除溶,剩余物用CH2Cl2萃取,无水硫酸镁干燥。旋蒸除溶后经中性氧化铝柱层析(洗脱剂:B)纯化得黄色固体7,产率60.9%;1H NMR δ:7.70~7.65(m,2H),7.54(d,J=7.0 Hz,2H),7.53(s,2H),7.36(s,4H),7.22(s,2H),7.12(d,J=5.3 Hz,2H),7.10(s,2H),4.10(s,4H),3.59(d,J=35.8 Hz,16H),3.36(s,4H),3.26(s,6H); MALDITOF-MS m/z:Calcd for C44H46N4O8{[M + H]+} 758.872 0,found 759.470 5。
2 结果与讨论
2.1合成
溴和一水合1,10-邻菲啰啉发生溴化反应可生成单溴代产物(1)和二溴代代产物(2)。通过控制溴的加量可提高1的产率。
合成6时,曾尝试以2,5-二碘对苯二酚为原料,先引入OTEG链再与双联频哪醇硼酸酯反应,但没有6生成。MALDI-TOF-MS显示只生成了6的二聚产物。换用反应活性较低的4为原料,后处理时通过GPC分离还得到二聚和三聚产物。
合成7的反应为Suzuki反应,对氧气比较敏感,因此反应体系需严格除氧。分离时,不宜使用硅胶柱层析,需采用中性氧化铝柱层析。此外,7在溶液中显示出强π┉π堆积作用,其1H NMR谱图需经升温测试获得。
7在THF等多种有机溶剂中溶解性良好,双邻菲啰啉结构为金属配位提供了更多的反应位点,从而在有机电致发光材料和超分子组装等领域有较好的应用前景。
参考文献
[1]Whitesides G M,Grzybowski B.Self-assembly at all scales[J].Science,2002,295(5564):2418-2421.
[2]陈华梅,岳凡,关晓梅.2-(2,4-二溴苯酚基)-1H-咪唑[4,5-f][1,10]邻菲罗啉的合成及其对阴离子的识别性能[J].合成化学,2010,18(5):567-571.
[3]Zhang S.Emerging biological materials through molecular self-assembly[J].Biotechnol Adv,2002,20(5):321-339.
[4]关磊,范文婷,王莹,等.新型单核Ni(Ⅱ)配合物{[Ni(phen)(H2O)4]·(1,5-nds)·H2O}的合成及其晶体结构[J].合成化学,2014,22(5):660-663.
[5]阎冰,张洪杰,王淑彬,等.稀土N-苯基邻氨基苯甲酸-1,10-邻菲罗啉二元、三元配合物的合成、表征及光物理性质[J].高等化学学报,1998,19(5):671-675.
[6]徐志栋,王敏,冯殿忠.稀土发光配合物的研究[J].应用化学,1999,16(2):18-22.
[7]李斌,张洪杰.双亲性稀土-邻苯二甲酸正十四醇单酯-邻菲罗啉三元配合物的合成、表征、荧光特性及成膜性能[J].无机化学学报,1998,14(2):233-236.
[8]李襄宏,唐定国.基于1,10-邻菲罗啉衍生物的两亲性钌配合物的合成及其光电转化性质[J].中南民族大学学报,2009,29(3):9-13.
[9]Frey J,Kraus T,Heitz V,et al.Synthesis of a bis-macrocycle containing two back-to-back rigidly connected 1,10-phenanthroline units as a central core and its incorporation in a handcuff-like catenane[J].Chemistry-A European Journal,2007,13:7584-7594.
[10]Sangvikar Y,Fischer K,Schmidt M,et al.Suzuki polycondensation with a hairpin monomer[J].Org Lett,2009,11(18):4112-4115.
[11]Traser S,Wittmeyer P,Rehahn M.Syntheses and solution properties of water-soluble poly(p-phenylene)s bearing oligo(ethylene oxide)and trialkylamino side groups[J].e-Polymers,2002,32:452-456.
[12]Strickler J H,Webb W W.Three-dimensional optical data storage in refractive media by two-photon point excitation[J].Opt Lett,1991,16(22):1780-1782.
·研究简报·
Synthesis of a Novel Phenanthroline Derivative
LI Rong1a,WANG Qiang1a,ZHANG Deng-qing1a,LI Xian-ying1b,XIANG Yun-jie1a,JIN Wu-song1a
(a.College of Chemistry,Chemical Engineering and Biotechnology; b.School of
Environmental Science and Engineering,1.Donghua University,Shanghai 201620,China)
Abstract:2,2'-【2,5-bis{ 2-[2-(2-methoxyethoxy)ethoxy]ethoxy}-1,4-phenylene】bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane(6)was prepared by a two-step reaction from 2-[2-(2-methoxyethoxy)ethyl]-4-methylbenzene sulfonate.A novel phenanthroline derivative,3,3'-【2,5-bis{ 2-[2-(2-methoxyethoxy)ethoxy]ethoxy}-1,4-phenylene】bis(1,10-phenanthrdin),was synthesized by Suzuki coupling reaction of 3-bromo-1,10-phenanthrdine with 6.The stucture was characterized by1H NMR and MALDI-TOF-MS.
Keywords:1,10-phenanthroline; Suzuki coupling reaction; synthesis
通讯作者:金武松,教授,博士生导师,Tel.021-67792391,E-mail:wsjin@ dhu.edu.cn
作者简介:李荣(1990-),男,汉族,江苏南通人,硕士研究生,主要从事功能有机分子的合成研究。E-mail:lirong1990@126.com
基金项目:国家自然科学基金资助项目(21172035,21302018)
收稿日期:2014-12-29;
修订日期:2015-09-17
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.12.1147 *
文献标识码:A
中图分类号:0625.15
