两种新型丁二炔衍生物的合成*
2016-01-17陈北华张灯青李贤英向芸颉金武松东华大学a化学化工与生物工程学院环境科学与工程学院上海201620
陈北华,张 振,张灯青,李贤英,向芸颉,金武松(1.东华大学a.化学化工与生物工程学院; b.环境科学与工程学院,上海 201620)
两种新型丁二炔衍生物的合成*
陈北华1a,张振1a,张灯青1a,李贤英1b,向芸颉1a,金武松1a
(1.东华大学a.化学化工与生物工程学院; b.环境科学与工程学院,上海201620)
摘要:以3,5-二溴-1-{ 3-(十二烷氧基)-2-[(十二烷氧基)甲基]丙氧基}苯和2-甲基-3-丁炔-2-醇为原料,经选择性Sonogashira偶联反应,Sonogashira偶联反应和去硅保护基反应制得中间体——3-乙炔基-5-(3-甲基-3-羟基)-丁炔基-1-(3-十二烷氧基)-2-{[(十二烷氧基)甲基]丙氧基}苯(6); 6经改良的Glaser偶联反应(CuI为催化剂,Et3N为溶剂)合成了一个新型的丁二炔衍生物(1)。6与2,2'-[(2,5-二碘-1,4-亚苯基)双(氧基)]双(四氢-2H-吡喃)经Sonogashira偶联,脱THP保护基和改良的Glaser偶联反应合成了一个新型的丁二炔衍生物(2)。中间体,1和2的结构经1H NMR,13C NMR和MALDI-TOF-MS表征。
关键词:丁二炔衍生物; Sonogashira偶联反应;改良Glaser偶联反应;合成
18818233161@163.com
通信联系人:金武松,教授,博士生导师,Tel.021-67792391,E-mail:wsjin@ dhu.edu.cn
二炔化合物作为构建分子的万能模块[1]而受到人们广泛的关注。丁二炔可作为许多官能团的等价物[2-3],其结构广泛存在于天然产物、抗真菌药物[4]中。最近它又成为纳米有机分子材料合成中的核心官能团[5];此外丁二炔类化合物也广泛应用于纳米科学[6]、分子转子[7]、液晶[8-11]、抗病毒活性研究[12]等领域,有广泛的研究价值和意义。
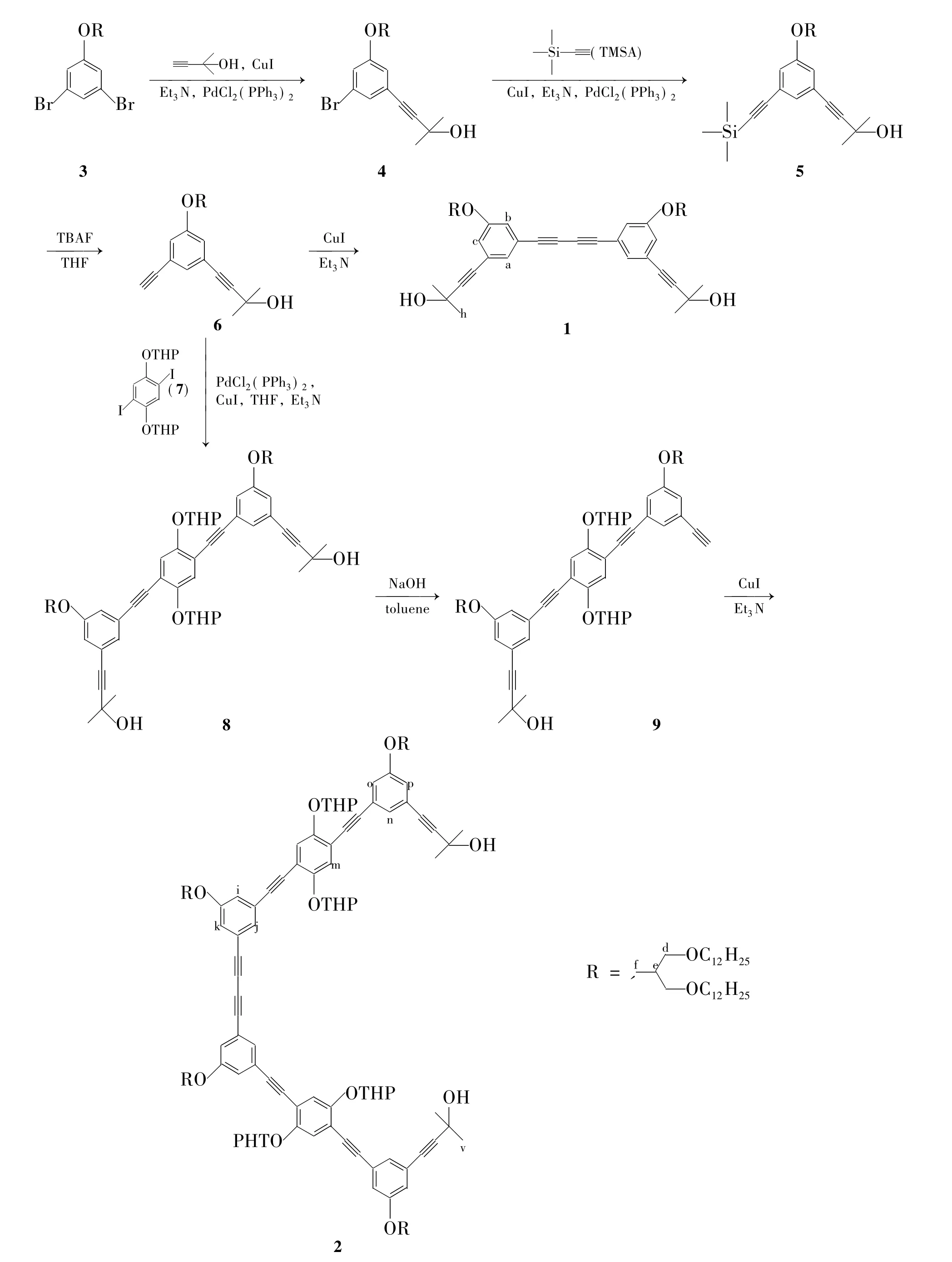
Scheme 1
在有机合成中,C-C的建立一直是最重要的模块也是难点之一。目前多采用金属有机试剂,丁二炔的合成采用sp-C间的连接,为形成此键,化学工作者尝试了多种方法[2,5,13-15]。但所用试剂多为重金属钯、钴催化剂、乙腈等价格昂贵、毒性大。且反应大多需要几种辅助试剂,反应成本高,操作繁琐,后处理复杂的同时也给环境带来了污染。
本文以3,5-二溴-1-{ 3-(十二烷氧基)-2-[(十二烷氧基)甲基]丙氧基}苯(3)和2-甲基-3-丁炔-2-醇为原料,经选择性Sonogashira偶联反应,Sonogashira偶联反应和去硅保护基反应制得中间体——3-乙炔基-5-(3-甲基-3-羟基)-丁炔基-1-(3-十二烷氧基)-2-{[(十二烷氧基)甲基]丙氧基}苯(6); 6经改良的Glaser偶联反应(CuI为催化剂,Et3N为溶剂)合成了一个新型的丁二炔衍生物(1)。6与2,2'-[(2,5-二碘-1,4-亚苯基)双(氧基)]双(四氢-2H-吡喃)(7)经Sonogashira偶联,脱THP保护基和改良的Glaser偶联反应合成了一个新型的丁二炔衍生物(2)(Scheme 1)。中间体,1和2的结构经1H NMR,13C NMR和MALDI-TOF-MS表征。
1 实验部分
1.1仪器与试剂
Bruker AV 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标); AB Sciex 4800型基质辅助激光电离飞行时间质谱(MALDI-TOF-MS)。
所用试剂均为分析纯。
1.2合成
(1)4的合成
在反应瓶中加入3 14.3 g(21.15 mmol),二(三苯基膦)二氯化钯99 mg和CuI 27.0 mg,氩气保护下抽换气3次;加入无水Et3N 120 mL,冷冻除氧3次。搅拌下用微量注射器注入2-甲基-3-丁炔-2-醇0.4 mL(4.23 mmol),于60℃(浴温)反应12 h。旋蒸除去Et3N,剩余物用CH2Cl2(3× 50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶层析柱(洗脱剂:二氯甲烷)纯化得白色液体4 2.5 g,产率89%;1H NMR δ:7.14(s,1H),7.04(s,1H),6.90(s,1H),4.02(d,J=5.7 Hz,2H),3.52(dd,J=5.8 Hz,1.6 Hz,4H),3.42(t,J=6.6 Hz,4H),2.36(p,J=5.9 Hz,1H),1.61(s,7H),1.28(d,J=10.7 Hz,43H),0.90(t,J=6.6 Hz,6H);13C NMR δ:159.50,126.68,125.09,122.34,118.45,116.44,94.76,80.72,77.36,77.04,76.73,71.42,68.65,66.70,65.44,53.38,40.01,31.94,31.39,29.69,29.66,29.65,29.50,29.37,26.19,22.70,14.12; MALDI-TOF-MS m/z:Calcd for C39H67O4BrNa{[M + Na]+}702.43,found 702.57。
(2)5的合成
在反应瓶中加入4 1.209 g(1.78 mmol),二(三苯基膦)二氯化钯62.5 mg和CuI 16.9 mg,氩气保护下抽换气3次;加入无水Et3N 80 mL,冷冻除氧3次。用注射器注入三甲基硅乙炔(TMSA)1.4 g(13.84 mmol),于60℃(浴温)反应24 h。旋蒸除去Et3N,剩余物用CH2Cl2(3×50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶层析柱[洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=10∶1]纯化得淡黄色液体5 1.1 g,产率89%;1H NMR δ:7.15(d,J=6.4 Hz,1H),7.06(s,1H),6.98(s,1H),6.93(d,J=11.3 Hz,1H),4.02(d,J=4.4 Hz,3H),3.53(d,J=5.7 Hz,5H),3.44~3.41(m,4H),2.36(q,J=5.9 Hz,1H),1.57(s,6H),1.27(s,51H),0.90(t,J=6.8 Hz,9H),0.26(s,9H);13C NMR(101 MHz,下同)δ:158.77,152.14,147.07,139.27,127.14,124.54,123.90,120.36,119.11,117.83,115.16,114.08,97.23,93.96,86.20,81.45,68.73,66.56,65.58,61.75,40.07,34.94,33.66,31.94,31.45,30.26,29.69,29.45,29.42,28.82,26.19,25.33,22.71,18.36,14.14; MALDI-TOF-MS m/z:Calcd for C44H76O4SiNa{[M + Na]+}719.55,found 719.68。
(3)6的合成
在反应瓶中加入5 1.116 g(1.64 mmol),氟化四丁基铵(TBAF)1.72 mg,氩气保护下抽换气3次;加入无水THF 40 mL,冷冻除氧3次,于室温反应12 h。用注射器注入饱和氯化铵溶液50 mL,搅拌5 min。用石油醚(3×50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶层析柱(洗脱剂:A=10∶1)纯化得淡黄色液体6 0.95 g,产率92%(直接投入下步反应)。1H NMR δ:7.15(s,1H),7.03(d,J=20.7 Hz,1H),6.95(d,J=25.7 Hz,1H),4.03(dd,J=5.7Hz,2.6 Hz,2H),3.54(d,J=6.0 Hz,5H),3.42(t,J=6.6 Hz,5H),3.06(s,1H),2.36(q,J=9.9 Hz,7.3 Hz,4.9 Hz,1H),1.56(q,J=6.9 Hz,8H),1.27(s,45H),0.90(t,J=6.7 Hz,8H); MALDI-TOF-MS m/z:Calcd for C41H68O4Na{[M + Na]+} 647.51,found 647.53。
(4)1的合成
在反应瓶中加入6 202.2 mg(0.324 mmol)和CuI 3 mg,搅拌下加入无水Et3N 50 mL,于60℃(浴温)反应48 h(TLC监测)。旋蒸除去Et3N,剩余物用二氯甲烷(3×50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶层析(洗脱剂:A=5∶1)纯化得淡黄色固体1 150 mg,产率74%;1H NMR δ:7.17(s,2H,a-H),7.01(d,J=10.9 Hz,4H,b,c-H),4.04(s,4H,d-H),3.54(dd,J=6.3 Hz,2.8 Hz,8H,f-H),3.42(td,J=6.8 Hz,3.0 Hz,8H,OCH2),2.37(d,J=5.7 Hz,2.5 Hz,2H,e-H),1.63(d,J=3.1 Hz,12H,h-H),1.40~1.19(m,95H,CH2),0.90(td,J=6.6 Hz,2.8 Hz,12H,CH3in R);13C NMR δ:158.76,128.02,124.15,122.75,119.05,118.49,94.38,81.08,80.80,73.95,71.42,68.68,66.61,65.53,40.05,31.93,31.41,29.69,29.65,29.50,29.37,26.19,22.69,14.12; MALDI-TOF-MS m/z:Calcd for C82H134O8Na{[M + Na]+}1 270.97,found 1 270.22。
(5)8的合成
在反应瓶中加入6 638.7 mg(1.022 mmol),7 254.3 mg(0.48 mmol),二(三苯基膦)二氯化钯26.9 mg及CuI 7.3 mg,氩气保护下抽换气3次;加入无水Et3N 50 mL,冷冻除氧3次;于60℃(浴温)反应48 h。旋蒸除Et3N,剩余物用CH2Cl2(3×50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶层析(洗脱剂:A=5:1)纯化得淡黄色液体8 500 mg,产率66.7%;1H NMR δ:7.27(s,2H),7.21~7.17(m,3H),7.03(dd,J=2.4 Hz,1.3 Hz,2H),6.97(dd,J=2.5 Hz,1.4 Hz,2H),5.53(p,J=3.3 Hz,2.6 Hz,2H),4.08~4.04(m,4H),3.55(d,J=6.0 Hz,9H),3.44(d,J=6.7 Hz,8H);13C NMR δ:158.77,152.14,147.07,139.27,127.14,124.54,123.90,120.36,119.11,117.83,115.16,114.08,97.23,93.96,86.20,81.45,68.73,66.56,65.58, 61.75,40.07,34.99,34.52,34.24,33.65,33.61,31.94,31.45,30.26,29.69,29.45,29.42,28.82,26.19,25.33,22.71,18.36,14.14; MALDI-TOF-MS m/z:Calcd for C98H154O12{[M-2THP]+} 1 355.03,found 1 355.04。
(6)9的合成
在反应瓶中加入8 200 mg(0.136 mmol)和NaOH 27.2 mg(5 eq.),氩气保护下抽换气3次;加入无水甲苯50 mL,冷冻除氧3次;回流(120 ℃)反应过夜(TLC监测)。用二氯甲烷(3×50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶层析(洗脱剂:A=8∶1)纯化得无色液体9 97 mg,产率50.2%; MALDI-TOF-MS m/z:Calcd forC95H148O11K{[M + K]+} 1 504.05,found 1 504.04。
(7)2的合成
在反应瓶中加入9 203.4 mg(0.144 mmol)和CuI 3 mg,搅拌下加入无水Et3N 50 mL,于60℃(浴温)反应48 h(TLC监测)。旋蒸除Et3N,剩余物用CH2Cl2(3×50 mL)萃取,合并有机相,用无水硫酸镁干燥;旋蒸除溶后经硅胶柱层析(洗脱剂:A=5∶1)纯化得淡黄色油状固体2 107.8 mg,产率53%;1H NMR δ:7.57(d,J=8.6 Hz,4H,m-H),7.23~7.07(m,8H,i,k,o,p-H),7.01(d,J=25.6 Hz,4H,j,n-H),5.54(s,4H,f-H),4.09~4.04(m,9H in THP),3.69(d,J=11.3 Hz,5H in THP),3.56(d,J=6.0 Hz,16H,d-H),3.44(td,J=6.5 Hz,2.9 Hz,16H,OCH2),2.39(q,J=13.2 Hz,7.3 Hz,6.8 Hz,4H,e-H),2.23~1.68(m,32H in THP),1.65(s,12H,v-H),1.29(d,J=13.6 Hz,235H,CH2),0.91(d,J=6.5 Hz,24H,CH3in R);13C NMR δ:158.81,152.17,147.07,139.26,127.77,12.14,124.90,124.53,124.46,123.98,123.91,97.38,97.23,86.58,86.19,81.44,80.94,73.99,71.44,68.72,66.57,65.57,61.76,40.09,34.88,34.53,33.84,31.95,31.45,30.34,30.20,29.69,29.52,29.38,29.18,28.97,18.37,14.14; MALDI-TOF-MS m/z:Calcd for C190H294O22Na{[M-4THP +Na]+}2 618.93,found 2 620.38。
2 结果讨论
4的合成是一个选择性的Sonogashira偶联反应,PdCl2(PPh3)2和CuI作为共催化剂,Et3N既作碱又作溶剂,同时也是缚酸剂,通过探索控制n(3)∶n(2-甲基-3-丁炔-2-醇)=5,于60℃反应12 h,可以得到高产率(89%)的单取代产物。
在9的合成中,NaOH需在研钵中研成粉末,通过探索控制NaOH的用量为5当量时,可以得到脱去一个保护基的产物。因为9在空气中不太稳定,故MS检测后直接投入下一步反应。
在1和2的合成中,本文以CuI为催化剂,这种方法不仅适合简单类丁二炔的合成也适合结构更复杂的丁二炔化合物的合成。该方法催化剂廉价易得、反应条件温和、过程简单易操作,产率也较高。
3 结论
本文通过简单的有机化学反应,成功地合成了两种新型的丁二炔类,为丁二炔类化合物的合成与物性研究提供了新的素材。
该方法具有反应条件温和、原料廉价易得、操作和后处理简单等优点,对合成丁二炔衍生物具有一定的参考价值。
参考文献
[1]Hunstman V D.The Chemistry of the Carbon-Carbon Triple Bond[M].London:Wiley-Interscience,1978.
[2]Valenti E,Pericas M A,Sarratosa F.1,4-Dialkoxy-1,3-but adiynes[J].J Am Chem Soc,1990,112:7405-7406.
[3]Siemsen P,Livingston R C,Diederich F.Acetylenic coupling:A powerful tool in molecular construction [J].Angew Chem Int Ed,2000,39:2632-2657.
[4]Stts A.A new class of active substances in antifungal chemotherapy[J].Angew Chem Int Ed Engl,1987,26:320-328.
[5]Xu Z,Kaha M,Walker K L,et al.Phenylacetylene dendrimers by the divergent,convergent,and doublestage convergent methods[J].J Am Chem Soc,1994,116:4537-4550.
[6]Michl J,Sykes E C H.Molecular rotors and motors:Recent advances and future challenges[J].ACS Nano,2009,3:1042-1048.
[7]Kottas G S,Clarke L I,Horinek D,et al.Artificial molecular rotors[J].Chem Rev,2005,105:1281-1376.
[8]Wu S T,Meng H B,Dalton L R.Diphenyl-diacetylene liquid crystals for electro-optic application[J].J Appl Phys,1991,70:3013-3017.
[9]Wu S T,Margerum J D,Meng H B,et al.Room-temperature diphenyl-diacetylene liquid crystals[J].Appl Phys Lett,1992,61:630-632.
[10]Wu S T,Neubert M E,Keast S S,et al.Wide nematic range alkenyl diphenyldiacetylene liquid crystals[J].Appl Phys Lett,2000,77:957-959.
[11]Beristain M F,Ogawa T,Gomez S G,et al.Polymerization of diphenylbutadiyne by gammarays irradiation in the molten state[J].Mol Cryst Liq Cryst,2010,521:237-245.
[12]Sari O,Roy V,Balzarini J,et al.Synthesis and antiviral evaluation of C5-substituted-(1,3-diyne)-2'-deoxyuridines[J].Eur J Med Chem,2012,53:220-228.
[13]Nishihara Y,Kegashira K,Hirabayashi K,et al.Coupling reactions of alkynylsilanes mediated by a Cu(I)salt:Novel syntheses of conjugate diynes and disubstituted ethynes[J].J Org Chem,2000,65:1780-1787.
[14]Damle S D,Seomoon D,Lee P H.Palladium-catalyzed homocoupling reaction of 1-iodoalkynes:A simple and efficient synthesis of symmetrical 1,3-diynes [J].J Org Chem,2003,68:7085-7087.
[15]Krafft M E,Hirosawa C,Dalal N.Cobalt-catalyzed homocoupling of terminal alkynes:Synthesis of 1,3-diynes[J].Tetrahedron Lett,2001,42:7733-7736.
·快递论文·
Synthesis of Two Novel Diacetylene Derivatives
CHEN Bei-hua1a,ZHANG Zhen1a,ZHANG Deng-qing1a,LI Xian-ying1b,XIANG Yun-jie1a,JIN Wu-song1a
(a.College of Chemistry,Chemical Engineering and Biotechnology; b.School of
Environmental Science and Engineering,1.Donghua University,Shanghai 201620,China)
Abstract:A novel diacetylene derivative(1)was synthesized by improved Glaser-coupling reaction(CuI as catalyst and Et3N as solvent)of 3-ethynyl-5-(3-methyl-3-hydroxyl)-butynyl-1-(3-dodecyloxy)-2-{[(dodecyloxy)methyl]propoxy} benzene(6),which was obtained by selective Sonogashira reaction,Sonogashira-coupling reaction and deprotection from 3,5-dibromo-1-{ 3-(dodecyloxy)-2-[(dodecyloxyyl)methyl]propoxy} benzene and 2-methyl-3-butyn-2-ol.A novel diacetylene derivative(2)was synthesized by Sonogashira reaction,THP deprotection reaction and improved Glaser-coupling reaction from 6 and 2,2'-[(2,5-diiodo-1,4-phenylene)bis(oxy)]bis(tetrahydro-2H-pyran).Structures of intermediate,1 and 2 were characterized by1H NMR,13C NMR and MALDI-TOF-MS.
Keywords:diacetylene derivative; selective Sonogashira reaction; improved Glaser-coupling reaction; synthesis
作者简介:陈北华(1987-),男,汉族,江西吉安人,硕士研究生,主要从事功能有机分子的合成与物性研究。E-mail:
基金项目:国家自然科学基金资助项目(21172035,21302018)
收稿日期:2014-12-26;
修订日期:2015-08-16
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.12.1130 *
文献标识码:A
中图分类号:O623.124; O621.3
