N掺杂BiPO4的第一性原理研究
2016-01-11李军奇崔明明刘振兴
李军奇, 袁 欢, 孙 龙, 崔明明, 刘振兴
(陕西科技大学 材料科学与工程学院, 陕西 西安 710021)
N掺杂BiPO4的第一性原理研究
李军奇, 袁欢, 孙龙, 崔明明, 刘振兴
(陕西科技大学 材料科学与工程学院, 陕西 西安710021)
摘要:采用了N原子对BiPO4进行掺杂.利用Materials Studio软件中的CASTEP模块,基于密度泛函理论的第一性原理,研究了N掺杂BiPO4的能带结构和电子态密度.结果表明,N掺杂导致BiPO4中出现杂质能级,禁带宽度变窄,电子迁移率有所改变.这对电子的捕获以及抑制电子/空穴的复合起到了很好的作用.
关键词:N掺杂; BiPO4; 第一性原理; 能带结构; 电子态密度
0引言
最近研究发现,对铋盐光催化剂进行掺杂能明显地增强光学吸收性能,能提高使用太阳光进行光催化反应的效率.通过控制磷酸盐半导体的形貌来优先暴露出光催化活性较高的晶面,有助于提高可见光照射下光催化剂的光催化活性[1].
Shang[2]发现N掺杂BiWO6在可见光降解RhB和CH3CHO时具有很好的表现.N掺杂BiPO4,在价带顶出现了杂质能级,从杂质能级到导带的直接电子跃迁能相对于纯净的BiPO4减小,从理论上解释了N掺杂BiPO4使吸收边际红移的实验现象,与Shang在实验中观察到的带隙减小趋势一致[1].同时,价带主要由O 2p和N 2p的杂化态组成,杂质使价带的宽度增加且没有出现隙态,这表明对光的吸收能力增加且光生载流子在价带中的迁移率提高[3].
经过前期研究发展,基于密度泛函理论的第一性原理已经成为研究原子、分子和固体电子结构的重要工具.利用第一性原理方法,对宽禁带半导体材料的电子结构、掺杂等进行计算机模拟,可以在相应计算结果的基础上对材料的物理性能进行预测和解释,从而进一步修正原有实验方法或进一步实现理论上的预测[4-7].
本文利用基于密度泛函理论的第一性原理研究了N掺杂BiPO4的能带结构、态密度、光吸收性质等,同时与纯净的BiPO4进行了对比,从而更好地了解了N在BiPO4内部的掺杂情况.
1模型建立与计算方法
1.1建立晶体模型
室温下,BiPO4的晶体结构[8]如图1所示.空间群为P21/n,单元晶胞参数为a=6.752 7 Å,b=6.943 0 Å,c=6.474 5 Å,β=103.71 °,α=γ=90 °,其中,PO是以PO4四面体形式存在,每个Bi原子周围有8个氧原子,晶体结构中离子键与共价键共存.这8个氧原子中分为两配位和三配位两种.
图1 未掺杂的BiPO4结构模型
在分析过程中,采用以下的局域轨道基作为价轨道:O(2s2,2p4)、P(3s2,3p3)、Bi(6s2,6p3)、N(2s2,2p3),平面截断能为300 eV,迭代收敛精度为2.0×10-6eV/atom,每个原子受力不超过0.005 eV/nm.
本文采用替位掺杂,通过在BiPO4的超晶胞中用杂质原子替位一个O原子进行模拟.为了减少边界效应的影响,选取两配位的O原子进行替换.在计算过程中,建立2×2×2的超晶胞结构,如图2所示.计算在不固定任意参数下进行几何优化,且在倒格矢空间进行,并设置为非自旋极化处理.

图2 沿晶胞a、b、c基矢量方向扩展两个单位后得到的2×2×2的BiPO4超晶胞
1.2计算方法
本文中的计算工作,应用Materials Studio中的CASTEP软件包完成.CASTEP[9]基于密度泛函理论,从头算的量子力学程序,利用平面波赝势方法,将离子势用赝势替代,电子波函数用平面波基矢组展开,电子-电子相互作用的交换和相关势由局域密度近似(LDA)或广义梯度近似(GGA)进行校正.这是目前较为准确的电子结构计算的理论方法[10,11].
由于所建立的晶体结构需要进行优化,对于优化方式的选择采用基于密度泛函的第一性原理能带计算方法进行量子计算.选用GGA/PBE优化方式,即交换关联函数采用广义梯度近似(GGA, Generalized Gradient Approximation)下的PBE(Perdew Burke and Ernzerhof)梯度修正函数;采用超软赝势描述价电子和原子实的相互作用.优化完成后,进行能量以及电子密度的模拟计算,最后得到电子能态密度和能带结构等数据及相关图表.
2结果与讨论
2.1BiPO4在参数设置下所得能带结构及电子态密度
2.1.1能带结构
图3为沿简约布里源区高对称点计算所得的本征BiPO4和N掺杂BiPO4的能带结构图.图3中零点位置为费米能级,本征BiPO4的价带处于费米能级之下,导带处于费米能级之上,其价带顶和导带底处在不同的对称点,或者说价带底和导带顶的跃迁没有出现在同一位置,是典型的间接带隙半导体能带结构.

(a)BiPO4 (b)N掺杂BiPO4图3 BiPO4与N掺杂BiPO4的能带结构图
在分析过程中,能带结构图与电子能态密度图相对应.能带结构图中曲线越密集,则电子能态密度图中相对应的能量位置峰值越大,表明该位置上的电子越多;反之,曲线越稀疏,则峰值越小,电子越少[12].在图3(a)中,电子在-18.72~ -17.76 eV和-2.84~ -0.28 eV范围内密度最大.
2.1.2电子能态密度
图4给出了BiPO4的总体电子态密度(TDOS)和各原子的分波态电子密度图(PDOS).从图4中可以看出,费米能级附近的峰主要是由O 2p和P 3p电子杂化组成.价带主要是由P 3s,O 2s和Bi 6s能级组成,其中O 2p和P 3p的杂化能级组成价带边,导带主要是由Bi 6p和P 3p能级组成,Bi 6p主要对导带底形成有贡献,但是对价带的贡献很小.
可以看出,BiPO4的能带位置为: -20.50~10.36 eV,带宽为30.86 eV.其中,电子在-18.87~ -17.06 eV和-2.67~ -1.62 eV范围密度最大,也就是说,电子在这两个区间数量最多;而电子在0.19~3.90 eV的电子密度非常小,几乎接近于0,这一区间的带宽为3.71 eV,与参考文献[13]中给出的3.85 eV非常相近.但理论计算值偏小于实验值,这是由于用第一性原理计算的禁带都普遍偏低的原因[14].
因为在计算过程中过高地估计了Bi 6s的电子能量,造成Bi 6s与O 2s电子相互作用增大,导致价带带宽增大,带隙偏低.为了克服计算方法带来的带隙低估并尽可能地符合实际情况,通常采用一种有效的修正方案“剪刀符算法”将半导体材料的导带位置上移,使得在计算其相关光学性质时尽可能与实验现象保持一致.
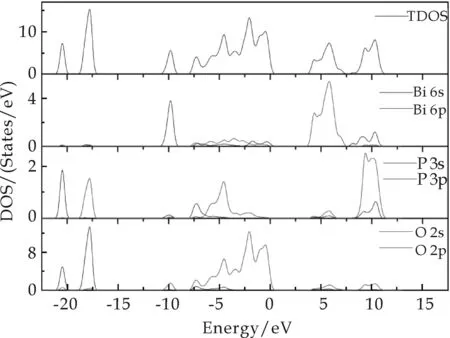
图4 BiPO4的总态密度(TDOS)和分态密度(PDOS)
2.2N掺杂的BiPO4在参数设置下所得能带结构及电子态密度
2.2.1能带结构
掺杂作为一种常见的提高光催化性能的有效方式,可使响应光吸收边际发生移动.在本文中,采用非金属元素N当作杂质原子引入使用.由图3(b)可知,在-19.48~-18.04 eV范围内,电子密度最大,电子数量最多.与本征结构相比,掺杂后电子分布更加局域.
在图3(b)中,掺杂态的能带带隙为3.29 eV,且从图中可以看出价带和导带位置发生微弱地上移.这是由于杂质态的引入,造成价带与导带之间有新的杂质能级生成,能带层数相应增多,变得更加致密.这些杂质能级作为中间能级,可以有效地协助电子从价带跃迁到导带,从而提高电子迁移率.
2.2.2电子态密度
掺入N原子的BiPO4结构发生了变化,从电子态密度的分布可以看出来.图5为N掺杂的BiPO4的总态密度(TDOS)和分态密度(PDOS).从图5分析可知,费米能级附近的峰值主要由P 3p和O 2p电子杂化组成.可注意到靠近费米能级处的P 3p的电子态密度峰值相对于O 2p电子密度明显较强,而且分布非常局域.价带主要是由Bi 6s和O 2s能级组成,其中P 3p和Bi 6p的杂化能级组成价带边,导带主要是由N 2p和Bi 6p能级组成.N 2p主要对导带形成有贡献,对价带的贡献很小.
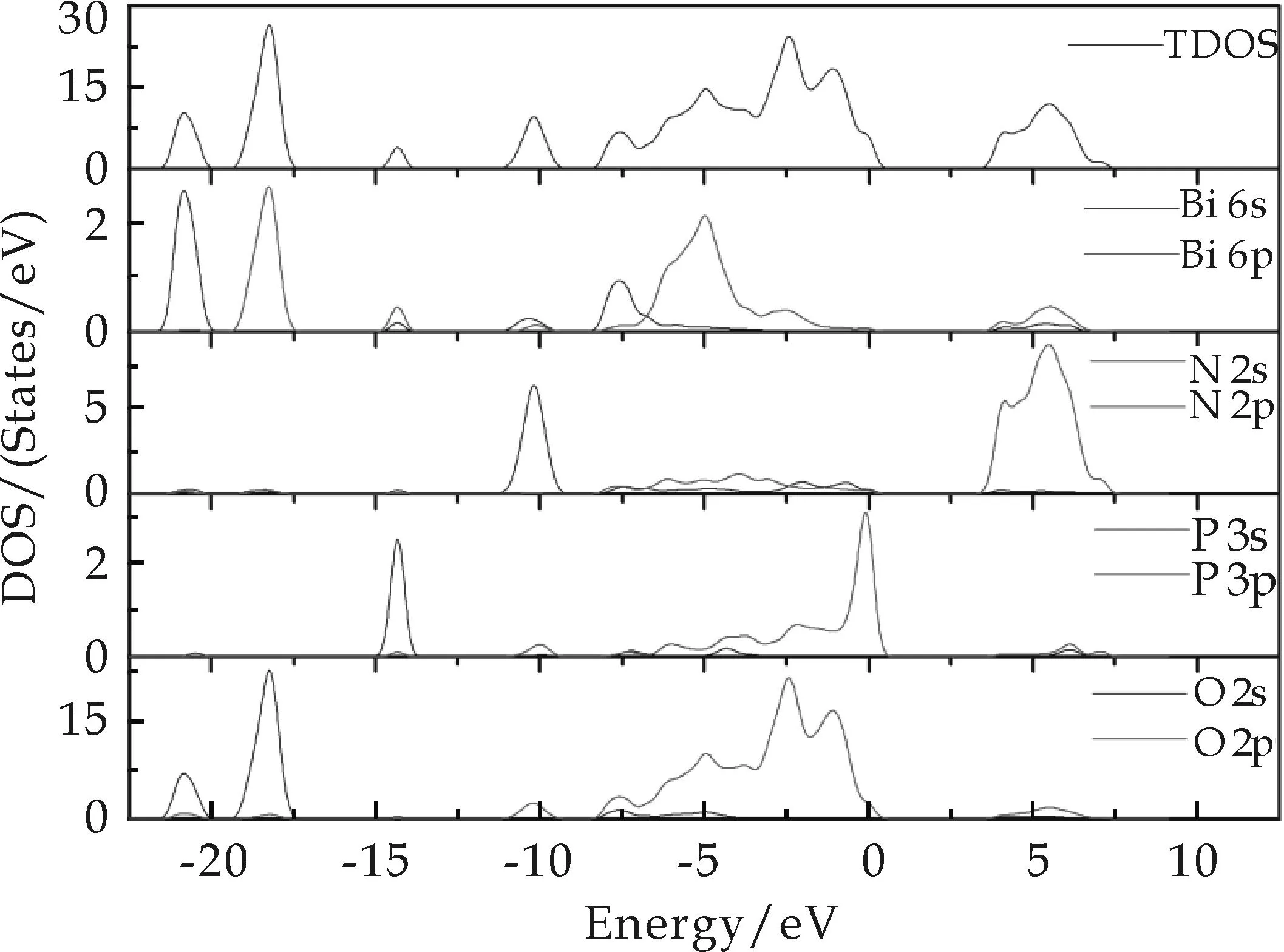
图5 N掺杂的BiPO4的总态密度(TDOS)和分态密度(PDOS)
N掺杂的BiPO4中的光诱导激发电子从Bi 6s和O 2s态跃迁至N 2p态.另一方面,N掺杂的BiPO4的较低价带区域(-7.8~-5.75 eV)是由Bi 6s和O 2p态杂化形成,使得该区域的能级分散度较大,光生空穴迁移能力变强,提高了BiPO4的氧化能力,使其表现出了良好的光催化性能.电子在0.33~3.62 eV范围内电子密度很小,几乎接近于0,这一区间带宽为3.29 eV.与未掺杂之前的3.71 eV相比,吸收边际发生明显红移,使用性能大大提高.
3结论
本文基于密度泛函理论下的第一性原理平面波超软赝势法,几何优化结构并计算了N掺杂后BiPO4的能带结构和电子态密度.
(1)掺杂后在导带与价带间形成杂质能级,此时的杂质能级为浅施主能级,捕获电子,延长了光生电子与空穴的复合时间,提高了电子和空穴的分离率.
(2)掺杂后禁带宽度减小,价带上的电子与未掺杂前相比获得较少的能量便可跃迁到导带.此外,由于杂质的引入,吸收光强变大,光吸收谱线范围有小幅增加,但仍处于紫外光范围.
参考文献
[1] 李军奇,郭占云,王德芳,等.立方体形Ag3PO4可见光光催化剂的制备及性能研究[J].陕西科技大学学报(自然科学版),2013,31(4):24-28.
[2] Shang M,Wang W Z,Zhang L,et al.Bi2WO6with significantly enhanced photocatalytic activities by nitrogen doping[J].Mater.Chem.Phys,2010,120(1):155-159.
[3] 赖康荣.几种铋基半导体材料的电子结构及光催化性质的理论研究[D].济南:山东大学,2012.
[4] 李青坤,王彪,王强,等.碳掺杂二氧化钛光催化性能的第一性原理研究[J].黑龙江大学学报(自然科学版),2007,24(4):455-457.
[5] 侯兴刚,刘安东,V+注入锐钛矿TiO2第一性原理研究[J].物理学报,2007,56(8):4 896-4 900.
[6] 赵宗彦,柳青菊,朱忠,等.锐钛矿相TiO2电子结构和光学性质的第一性原理计算[J].半导体学报,2007,28(10):1 555-1 561.
[7] Cristiana D V,Emanuelef,Glanfranco P,et al.N-doped TiO2theory and experiment[J].Chemical Physics,2007,339(4):44-56.
[8] 潘成思.BiPO4含氧酸盐新型光催化剂的可控合成及构效关系研究[D].北京:清华大学,2011.
[9] Segall M D,Lindan P J D,Probert M J.First-principles simulation:ideas,illustrations and the CASTEP code[J].Phys.Cond.Matt,2002,14:2 717-2 744.
[10] Keiji W,Masatoshi S,Hideaki T.Formation of composite oxide films on aluminum by Sol-Gel coating and anodizing for the development of high performance aluminum electrolytic capacitors[J].Electro chemistry,2001,69:407-434.
[11] 李震宇.新材料物性的第一性原理研究[D].合肥:中国科学技术大学,2004.
[12] 窦俊青,康雪雅,吐尔迪·吾买尔,等.Mn掺杂LiFePO4的第一性原理研究[J].物理学报,2012,61(8):341-348.
[13] Siyuan Wu,Hong Zheng,Youwei Lian,et al.Preparation,characterization and enhanced visible-light photocatalytic activities of BiPO4/BiVO4composites[J].Materials Research Bulletin,2013,48(8):2 901-2 907.
[14] Wei S H,Alex Zunger.Role of metal d states in Ⅱ-Ⅵsemieonduetors[J].Phy.Rev.B,1988,37:8 958-8 963.

First-principles research of nitrogen-doped BiPO4
LI Jun-qi, YUAN Huan, SUN Long, CUI Ming-ming, LIU Zhen-xing
(School of Materials Science and Engineering, Shaanxi University of Science & Technology, Xi′an 710021, China)
Abstract:In this article,the band structure and electronic states density of nitrogen-doped BiPO4were calculated by the CASTEP module of Materials Studio software according to the density functional theory.The study showed that N doping leads to the appearance of impurity level,smaller band gap and the mobility of electrons change when comparing with pure BiPO4.It actively affected the capture of internal electron and the inhibition of electronic/hole combination.
Key words:nitrogen-doped; BiPO4; first-principles; band structure; electronic density of states
中图分类号:O64
文献标志码:A
文章编号:1000-5811(2015)02-0056-04
作者简介:李军奇(1978-),男,陕西西安人,副教授,博士,研究方向:环境催化材料
基金项目:国家自然科学基金项目(51203136); 陕西科技大学学术骨干培育计划项目(XSGP201202); 陕西科技大学自然科学基金项目(ZX11-14)
收稿日期:*2014-12-06
