氧化物阳极在不同温度下硫酸及海水中强化寿命规律及机理分析
2015-12-31张胜健辛永磊
张胜健, 辛永磊
(1.太原工业学院化学与化工学院,山西 太原 030008;2.中国船舶重工集团公司第七二五研究所,海洋腐蚀与防护重点实验室,山东 青岛 266101)
钌、铱、锡阳极作为典型的析氯阳极被广泛地应用在电解海水防污、污水处理、海水淡化利用等多方面[1-3]。金属氧化物阳极的寿命是考察金属氧化物阳极性能的重要指标。金属氧化物阳极的寿命通常以强化电解寿命获得,即在大电流密度、苛刻电解质中强行运转[4]。温度是影响金属氧化物阳极寿命的一个重要因素,但钌、铱、锡阳极在实际应用与实验考察中阳极寿命会表现出不相符的规律。本文考察不同溶液中温度对电极寿命的影响,研究并对比钌、铱、锡阳极在海水中与硫酸中2种不同介质的失效机理,为改进钌、铱、锡类析氯电极的强化电解寿命测试准确性提供参考。
1 实验部分
1.1 氧化物阳极的制备
将工业用TA2钛板进行喷砂处理,除去表面氧化物,放入固定配比的Na2CO3、NaOH、NaH2PO4溶液中进行除油,10%草酸煮沸溶液中刻蚀约2h,成均匀的麻灰面,蒸馏水冲洗后备用。按照n(Ru)∶n(Ir)∶n(Sn)=17∶23∶60的比例配制涂液,均匀涂刷在处理好的钛基体上,120℃条件下烘干10min,马弗炉470℃下烧结10min,连续涂刷4层和12层,最后一次烧结1h,最终得到氧化物涂覆量为2.013g/m2和6.113g/m2的金属氧化物阳极。
1.2 电化学性能测试
强化电解寿命实验:溶液介质分别为1mol/L H2SO4和海水,温度为5、10、15、20、40℃。以钛板为阴极,以制备的钌、铱、锡金属氧化物电极为阳极,两极板间距1cm。在海水中,阳极电流密度为0.5A/cm2,1日换2次海水,槽压迅速升高到14V时认为失效,停止测试;硫酸中,强化电流密度为2A/cm2,当槽压迅速升高到14V时阳极失效,停止测试。
电化学阻抗测试采用M2273电化学工作站,选用三电极体系,铂铌丝为对电极,饱和甘汞电极为参比电极,1mol/L硫酸和3.5%NaCl溶液为电解液。在硫酸中外加测试电压为1.20V,3.5%的NaCl溶液中外加测试电压为1.13V,扫描频率均为100kHz~10MHz,电位扰动信号振幅为10mV。
1.3 阳极涂层形貌观察
采用PHILIP XL30扫描电镜对制备的金属氧化物进行表面形貌观察,并通过附带能谱仪对表面进行成分分析。
1.4 XRD物相结构分析
D8Advance衍射仪,采用钴靶,工作电压40kV,电流40mA。
2 结果与分析
2.1 阳极在2种溶液中强化电解寿命测试
图1示出了金属氧化物阳极在硫酸和海水中强化电解寿命随温度的变化。从图1中可以看出,金属氧化物阳极在2种溶液中随着温度的变化,强化电解寿命呈现出相反的趋势。在硫酸中,随着温度的上升,金属氧化物阳极的寿命减少,但减少幅度不大,这一结果与张招贤等的测试规律类似[4];在海水中,随着温度的上升,金属氧化物的强化电解寿命明显增长,其在5℃与40℃下强化电解寿命相差极大。这可能是由于,在2种溶液中的金属氧化物阳极表面析出气体不同,最终导致阳极的失效机理不同[5]。金属氧化物阳极在硫酸中主要发生析氧反应,在海水中主要发生析氯反应,导致两者的失效机制不同。温度会影响到金属氧化物阳极的表面活性点的数目、活化能以及反应速率[6-7]。温度越高,阳极表面析氧越快,负载的电催化活性物质剥落越快,同时Ru的溶解越快,导致高温溶液中金属氧化物阳极越快失效。对于在海水中的析氯反应,温度越高,电流效率越高,氧气的析出越少,对于有效物质Ru的溶解越少。在不同的溶液中阳极强化电解失效后,再在3.5%的NaCl溶液中进行电流效率的测试发现,各失效阳极的电流效率约为16.7%~21.4%,表明失效后的金属氧化物阳极仍然有较大的活性;而在硫酸中失效的阳极,电流效率极低,基本上已失活性,这与EDX分析结果一致。
2.2 表面形貌变化
图2a)~c)依次为新鲜试样、硫酸中失效试样、5℃海水中失效试样的SEM照片。除40℃失效阳极,其他失效阳极与5℃条件下失效阳极的表面形貌基本相似,故不列出。不同温度下硫酸中失效试样的SEM照片类似,故仅列出20℃下失效试样。表1为不同阳极的能谱元素分析表。
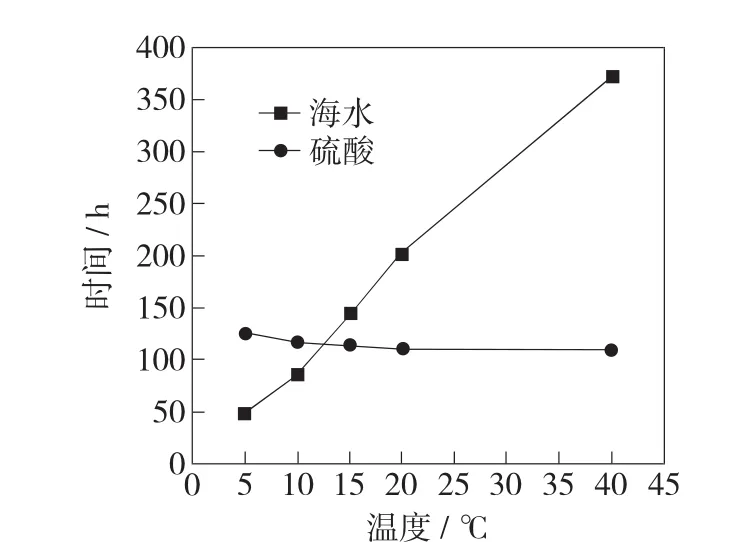
图1 金属氧化物阳极强化电解寿命随海水和硫酸温度变化曲线图
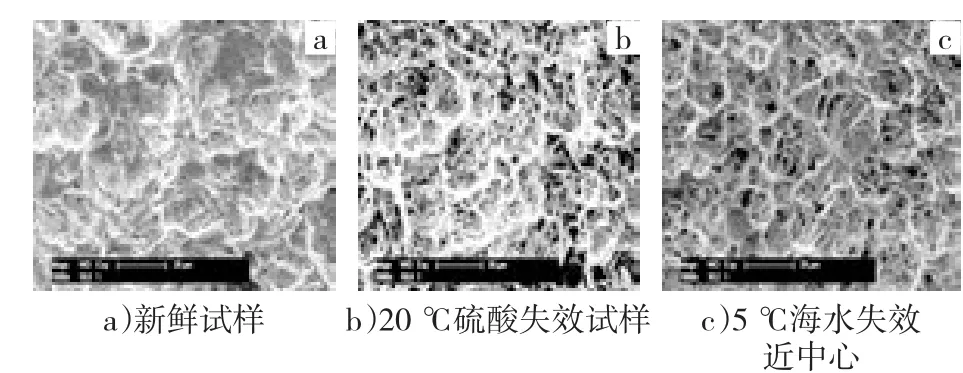
图2 不同金属氧化物阳极的SEM形貌(×1 000)
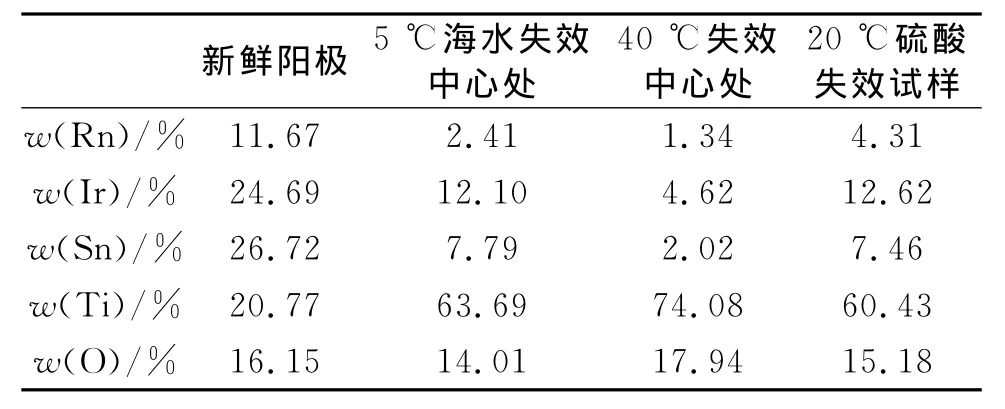
表1 不同阳极的能谱元素分析表
从图2中可以对比看出金属氧化物阳极在硫酸和海水中强化电解失效前、后表面形貌的变化。新鲜试样阳极表面有许多窄而浅的微裂纹,附着有微小结晶粒。在硫酸中进行强化电解,随着电解的进行,金属氧化物阳极表面的微观形貌并没有发生大的改变。经EDX分析发现,表面物质Ru较少。阳极强化电解失效后,金属氧化物阳极表面形貌完全改变,呈刻蚀后钛基体的蜂窝状。经EDX分析发现,表面的有效元素Ru、Ir、Sn均明显减少,但仍有少量的残余,O略有升高,Ti的含量迅速升高,表明钛基体基本裸露。阳极在海水中的失效呈现出2种不同的形貌变化。在温度较低时,氧化物阳极表面呈现出明显的分界,分界两侧表面形貌对比明显。经过EDX分析发现,两侧元素含量相差也较大,在靠近中心处,Ru、Ir、Sn虽有减少,但表面仍有较多残余。在40℃失效后,阳极表面形貌单一,与在硫酸中失效类似,呈现出基体的蜂窝状,涂层发生电化学溶解,有效成分Ru、Ir、Sn明显减少,钛基体基本裸露。
综合以上分析表明,金属氧化物阳极电催化成分在不同溶液中、不同温度下的电化学溶解及剥落方式影响其寿命。
2.3 电化学阻抗谱
用Rs(QdlRct)(QfRf)能够较好地表示等效电路,如图3所示。Rs(QfRf)拟合在40℃海水及在硫酸中失效阳极等效电路效果更好。阳极在硫酸中失效后的等效电路参数值相差不大,故仅列出20℃硫酸中失效的等效电路参数值。其中,Rs为溶液电阻,其值受温度、盐桥与阳极间距影响,Rs的值受阳极析气效应的影响。QfRf主要反应涂层的内表面与钛基体之间的物理性质,RctQdl反应涂层外表面与溶液界面的阻抗,即双电层阻抗。
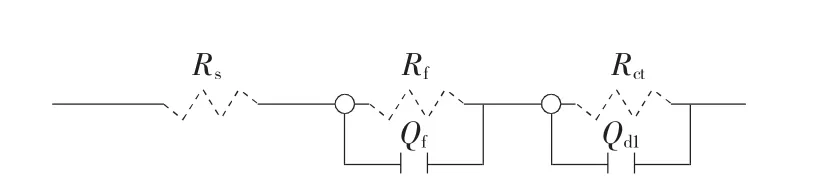
图3 阳极的等效电路图
采用Zismpwin软件对得到的阻抗谱图进行拟合分析,模拟电路各参数值如表2所示。
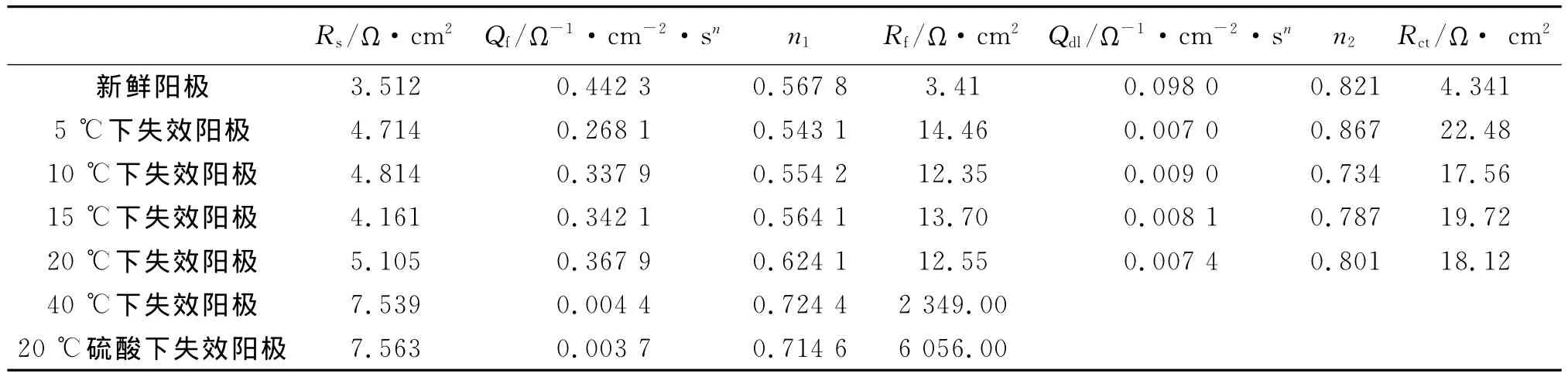
表2 不同电解温度失效Ru-Ir-Sn氧化物阳极的交流阻抗等效电路参数值
表2是金属氧化物阳极在不同温度、不同溶液中失效前、后的交流阻抗等效电路参数拟合值。阳极在不同硫酸中失效后各参数拟合值相差不大,故仅在表2中列出20℃下阳极的拟合参数值。从表2可以看出,新鲜阳极的Rf值与Rct值相对较小,在低温、室温海水中失效后有较大程度的增长,约为原来的4倍~5倍。结合表面形貌分析,可能是由于RuO2溶解以及电流的“边缘效应”、边缘处涂层有效物质脱落引起。在40℃海水中失效后,在阳极涂层表面的有效物质已很少,涂层与基体的电阻Rf可达2 349Ω·cm2。结合表面形貌分析,这是由于在涂层发生均匀性溶解、最终导致与基体之间生成不导电的TiO2钝化膜引起的。阳极在硫酸中失效后,涂层与基体的电阻Rf高达6 056Ω·cm2。经EDX分析发现,Ti基体基本裸露,含量达75%左右,氧的含量也升高,这是氧化物阳极在硫酸中电解后期电压迅速升高、快速失效的根本原因[8]。
2.4 失效机理对比分析
钌、铱、锡金属氧化物阳极在进行强化电解过程中,阳极表面会发生式(1)~式(4)电化学反应[9-10]。
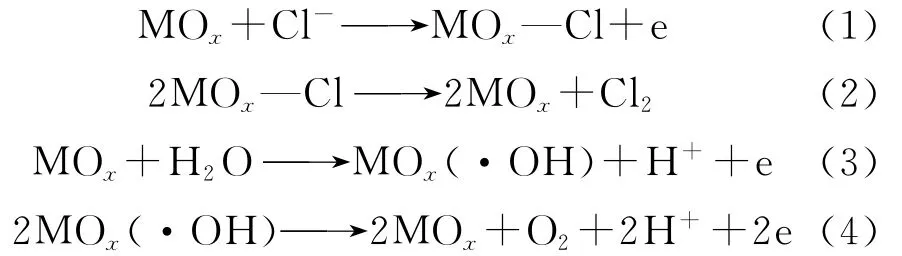
氧化物阳极在硫酸中进行强化电解阳极表面主要发生式(3)、(4)反应。研究表明,当阳极电位较高时,阳极表面会发生不可逆的电化学反应,RuO2会被氧化成水中可溶性的Ru,发生电化学溶解[11]。同时,电解产生的大量气泡冲击会导致涂层剥离,涂层的结构稳定性遭到破坏。随着电解的进行,产生的绝大部分氧气会进入到溶液中,微量的氧气通过扩散与迁移到达钛基体,与钛基体结合,发生相应的反应,产生高电阻的二氧化钛。随着电解的继续进行,TiO2生产不断增多[12],电压不断升高,最终导致阳极的失效。硫酸温度越高,析氧电位越低,析氧越容易,同时氧气的析出速率越快,金属氧化物阳极表面的RuO2溶解越快,涂层的剥离也越快,表现出阳极强化电解寿命随着硫酸的温度升高而降低。
阳极在海水中进行电解,在阳极表面会发生式(1)、(2)、(3)、(4)式反应。温度较低时,受到电流的“边缘效应”影响较大,边缘处的电流密度较大,边缘处容易发生析氧反应,阳极表面有效物质氧化物固溶体的溶解速度比中心处快,导致分界现象的产生。随着电解的进行,靠近中心处的有效面积越来越小,导致电压逐渐升高,最终失效;随着海水温度的升高,氯离子的扩散速率增快,析氧电位降低。因此,氧气的析出及RuO2电化学溶解的减慢,阳极表面发生均匀的电化学溶解。随着电解的进行,RuO2的电化学溶解增多,析氧增多,氧气吸附在钛基体上,产生高电阻的TiO2[8],最终导致电解的失效。海水温度升高,析氯效率增加,是金属氧化物阳极强化电解寿命随海水温度升高而降低的原因。
3 结论
1)金属氧化物阳极在硫酸与海水中寿命随着温度的升高表现出相反的规律。在海水中,随着温度的升高,氧化物阳极强化电解寿命增长;在硫酸中,随着温度的上升,阳极强化电解寿命缩短。钌、铱、锡类金属氧化物氧化在硫酸中进行强化寿命实验,可能会与实际寿命表现差距较大。
2)氧化物阳极在硫酸中与海水中的失效机制不同。在硫酸中,金属氧化物阳极的失效主要是阳极表面发生析氧反应,在钛基体上生产不导电的二氧化钛;在海水中的失效低温主要是发生析氯反应,失效由涂层的电化学溶解与剥落引起,在40℃下的失效是电化学溶解与生产二氧化钛,生产不导电的二氧化钛是失效的主要原因。
[1] 陈德斌,陈学群,孔小东,等.电解海水防污系统失效阳极研究[J].材料保护,2006,39(9):59-61.
[2] 孟惠民.贵金属氧化物电极电解处理有机废水[J].北京科技大学学报,2003,25(5):405-409.
[3] 王彬,侯世忠,韩严,等.海水电解防污用金属氧化物涂层阳极研究现状[J].材料开发与应用,1998(1):41-45.
[4] 张招贤,郑团.涂层钛阳极强化寿命试验电解因素的选择[J].氯碱工业,2004(5):10-11.
[5] 张胜健,杜爱玲,许立坤,等.海水温度对金属氧化物阳极强化电解失效行为影响[J].稀有金属材料与工程,2013,42(12):2613-2618.
[6] 唐 益,许立坤,王均涛,等.Ti/IrO2-Ta2O5-SnO2纳米氧化物阳极的研究[J].稀有金属材料与工程,2010,39(4):687-691.
[7] 初立英,许立坤,吴连波,等.草酸浸蚀对氧化物阳极形貌及电催化性能的影响[J].金属学报,2005,41(7):763-768.
[8] 黄运涛,彭乔.海水电解用金属氧化物阳极的失活机理[J].稀有金属材料与工程,2006,35(10):1610-1615.
[9] 张招贤.IrTa氧化物涂层钛阳极恶化原因分析[J].氯碱工业,2005(1):12-16.
[10]Tomcsányi L,De Battisti A,Hirschberg G,et al.The study of the electrooxidation of chloride at RuO2/TiO2electrode using CV and radiotracer techniques and evaluating by electrochemical kinetic simulation methods[J].Electrochimica Acta,1999,44(14):2463-2472.
[11]张琼,彭俊华,蔡传荣.钛阳极涂层剥落失效机理初探[J].电子显微学报,2001,20(4):410-411.
[12]王均涛,韩严,许立坤,等.Ru-Ir-Ti氧化物阳极正反电流电解失效机理研究[J].电化学,2005,11(4):407-411.
