碳化硅衍生碳/球形天然石墨复合材料的制备及其结构调控
2015-12-29杜雪莲李轩科崔正威董志军袁观明武汉科技大学省部共建耐火材料与冶金国家重点实验室武汉43008武汉科技大学湖北省煤转化与新型炭材料重点实验室武汉43008
杜雪莲 丛 野,* 姜 露 李轩科,* 崔正威董志军 袁观明 张 江(武汉科技大学,省部共建耐火材料与冶金国家重点实验室,武汉43008; 武汉科技大学,湖北省煤转化与新型炭材料重点实验室,武汉43008)
碳化硅衍生碳/球形天然石墨复合材料的制备及其结构调控
杜雪莲1,2丛 野1,2,*姜 露2李轩科1,2,*崔正威2董志军2袁观明2张 江2
(1武汉科技大学,省部共建耐火材料与冶金国家重点实验室,武汉430081;2武汉科技大学,湖北省煤转化与新型炭材料重点实验室,武汉430081)
为满足储能领域对于材料兼具高能量密度和高功率密度的需求,本文旨在将具有特殊孔隙结构的碳化物衍生碳与具有高导电性和高能量存储密度的石墨化碳(球形天然石墨)相复合,制备得到一种多孔碳化硅衍生碳/球形天然石墨(SiC-CDCs@NG)复合材料.采用X射线衍射(XRD)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、拉曼光谱、N2吸/脱附等方法对材料的组成、结构、形貌、孔结构和比表面积等进行了表征.结果表明, SiC-CDCs@NG材料具有较大的且可调节的比表面积和微孔体积,微孔孔径集中在0.5-0.7 nm范围内;通过改变NG/Si摩尔比,可以有效调控CDCs壳和NG核在复合材料中的组成分布、CDCs微孔的体积、孔径分布和比表面积.
碳化硅衍生碳;复合材料;结构调控;比表面积;孔径分布
©Editorial office ofActa Physico-Chimica Sinica
1 引言
碳化物衍生碳(CDCs)是以碳化物晶格为模板,通过选择性刻蚀将碳化物中的金属或非金属元素去除,并将碳骨架完好地保留而制得的纳米孔炭材料,1具有比表面积高、纳米孔结构发达、孔径分布窄且精确可调等优点,在双电层电容器、催化剂载体、气体储存等领域具有独特的应用优势.2-6
多孔CDCs的制备通常采用高温氯化法,用氯气刻蚀金属碳化物方法简单,CDCs膜厚容易控制且制备的产物纯净.7-10碳化物衍生碳的结构是其模板碳化物结构的复制,因此CDCs的结构首先取决于作为模板剂的碳化物的种类.如文献11中报道的用碳化钒(VC)和碳化铪(HfC)作碳化物前驱体比用碳化钛(TiC)、碳化锆(ZrC)、碳化钽(TaC)、碳化铌(NbC)作前驱体能制备更加有序的碳.也有报道表明微孔率和中孔率受起始碳化物前驱体及其密度的影响.12由于前驱体的差异,CDCs的孔隙率在50%-80%之间不等.CDCs的孔结构还可通过控制温度、时间、压强、刻蚀气体的流量和其他过程参数进一步调节,11-13其中温度的影响最为显著.随着氯化温度的升高,CDCs的结构将经历一个渐进向部分石墨化演化的过程,14-19600°C以下得到的CDCs主要是无定形结构,800°C左右开始出现一些短小、弯曲的石墨烯片结构,1200°C开始形成一些有序性好、厚1 nm至数纳米的石墨带、带状碳和晶须.14据文献报道,SiC在900-1000°C氯化可得到由无定形碳包围的纳米金刚石.13,14当提高氯化温度,在SiC/C界面处纳米金刚石会转化生成洋葱碳和石墨.15
单纯以碳化物衍生碳作为电极活性物质的超级电容器能表现出较高的比电容特性,20-24但仍主要依靠衍生碳大的比表面积和特殊的孔结构在电极表面发生物理吸附进行储能,和一般锂离子电池相比(比能量120-170 Wh·kg-1),能量密度不高(~10.8 Wh·kg-1).因此,我们设计了一种以多孔质碳化物衍生碳与具有高导电性和高能量储存密度的天然石墨(NG)相复合的材料作为储能用电极材料,期望该复合材料既具有超级电容器用材料合适的比表面积与孔隙结构进行双电层储能,又具有优异的锂离子脱/嵌可逆性,从而满足储能领域对于材料兼具高能量密度和高功率密度的需求.
本文以球形天然石墨和硅为原料通过固相反应生成碳化硅@球形天然石墨(SiC@NG)前驱体,然后通过氯气刻蚀制备了碳化硅衍生碳/球形天然石墨(SiC-CDCs@NG)复合材料,采用X射线衍射(XRD)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、拉曼光谱、N2吸/脱附对材料的组成、结构、形貌进行了表征.重点研究了球形天然石墨与硅(NG/Si)的摩尔比对SiC-CDCs@NG复合材料的组成、微观结构和形貌、孔结构及比表面积的影响.
2 实验部分
2.1 固相法合成SiC@NG前驱体
以球形天然石墨和硅粉为原料(NG规格:灰分0.1%,粒径15-20 μm,深圳市斯诺实业发展有限公司;Si粉:300目,纯度99.99%,北京中金研新材料科技有限公司),采用固相法在1600°C反应2 h(NG/Si摩尔比值分别为1、2、3、4),制备SiC@NG-R(R表示不同的NG/Si摩尔比)前驱体.
2.2 氯化法制备多孔SiC-CDCs@NG复合材料
将2.1节所制备的SiC@NG前驱体粉末样平铺在石英容器中,放入管式炉中,通入氩气,以5°C· min-1的升温速率加热至目标温度(1000°C),开始向管式炉中通入氯气,氯气流量为20-30 mL·min-1,并保持1 h.氯化完成后继续通氩气,将样品中残留的氯气和样品孔隙里生成的副产品带走,直到温度降到室温.反应生成的气态副产品用NaOH溶液吸收.
2.3 样品表征
XRD采用Philips X Pert MPD Pro型转靶X射线衍射仪,以阳极Cu靶,Kα辐射线(λ=0.154056 nm)为辐射源,测试管电压为40 kV,管电流为30 mA,扫描速率为8(°)·min-1,扫描范围为10°-90°.SEM采用VEGE 3 SBH-EasyProbe型扫描电子显微镜对样品进行测试.TEM采用日本电子公司的JEM-2100型透射电子显微镜,测试电压为200 kV.拉曼光谱分析采用Thermo公司RenishawinVia Reflex型激光显微拉曼光谱仪,使用氦氖激发器,激发波长为632.8 nm.N2吸附/脱附等温曲线采用美国Micromeritics公司的ASAP 2020物理吸附仪于77 K条件下测试,测试前,样品在真空条件下于350°C预先脱气4 h.样品的比表面积采用Brunauer-Emmett-Teller(BET)方法,根据相对压力(p/p0)在0.04-0.2范围内的吸附数据进行计算;孔径分布(PD)由等温线吸附分支采用密度泛函理论(DFT)模型计算,孔体积(孔容)用相对压力(p/p0=0.998)处的吸附量计算.
3 结果与讨论
3.1 SiC@NG前驱体结构和形貌表征
以球形天然石墨和Si粉为原料,采用固相法在1600°C反应2 h,制备了SiC@NG前驱体.图1是不同NG/Si摩尔比值(1、2、3、4)条件下所制备的SiC@NG前驱体的XRD图.从图中可以看出,在衍射角2θ=26.6°,42.3°,44.5°,54.8°,77.4°附近出现了几个较强的衍射峰,分别对应于碳的(002)、(010)、(011)、(004)、(110)晶面衍射峰(JCPDS:00-008-0415);在2θ=35.7°,41.5°,60.1°,71.9°附近出现了四个较强衍射峰,与立方相碳化硅(β-SiC)的标准谱图(JCPDS:01-089-8487)相比分别对应SiC的(111)、(200)、(220)、(311)晶面,表明在产物中主要存在未完全反应的碳(球形天然石墨)和固相反应生成的立方相SiC两相共存,没有其他杂质;随着NG/Si摩尔比的减小,碳的衍射峰强度明显减弱,当摩尔比值为1时,碳的衍射峰基本消失,而立方相SiC的衍射峰则明显增强,这表明NG与Si反应生成的SiC增多,因此可以通过改变NG/Si的摩尔比来控制固相反应的深度,调控SiC@NG前驱体中壳层SiC的厚度和NG核的尺度,并最终调控其刻蚀产物CDCs@NG中CDCs和NG的组成.

图1 不同NG/Si摩尔比值(R)条件下制备的SiC@NG前驱体的XRD图Fig.1 XRD patterns of precursor SiC@NG synthesized with different NG/Si molar ratios(R)
图2是NG原料和SiC@NG-2(NG/Si摩尔比为2)前驱体的SEM图及SiC@NG-2的能量色散X射线能谱(EDS)图.从图中可以看出,固相反应后球形天然石墨表面的粗糙程度明显增大,表面被包覆了一层由细小的颗粒聚集而成的新相,球体尺寸也有明显的增大,但球形结构基本保持不变.根据SiC@NG-2的EDS结果及结合XRD结果判断,球形天然石墨表面包覆层为与Si反应生成的SiC.因此,球形天然石墨与Si粉经1600°C固相反应后可以制备得到表面包覆均匀性较好的SiC涂层.

图2 (a)球形天然石墨原料和(b,c)SiC@NG-2的SEM图及(d)SiC@NG-2的EDS图Fig.2 SEM images of(a)NG and(b,c)SiC@NG-2,and(d)energy dispersive X-ray spectroscopy(EDS)image of SiC@NG-2
3.2 多孔SiC-CDCs@NG复合材料的结构表征
以氯气为刻蚀剂,将SiC@NG前驱体在1000°C氯化1 h.图3是不同NG/Si摩尔比值(1、2、3、4)条件下所制备的SiC-CDCs@NG的XRD图.从图中可以看出,当NG/Si摩尔比为2、3、4时,刻蚀后,产物中只有碳的衍射峰存在,而SiC的衍射峰已完全消失,这是因为SiC中的Si原子被氯气刻蚀掉而生成了新的衍生碳,且随着NG/Si摩尔比的增大,碳的(002)峰逐渐增强,这与图1中的结果相一致.而NG/Si摩尔比为1的SiC@NG前驱体刻蚀后仍然存在SiC的衍射峰,其原因是NG/Si摩尔比降低后,经固相反应生成的立方相SiC较多,在同样条件下,SiC不能完全反应.因此,对于含SiC较多的SiC@NG前驱体需要通过增加氯气流量、提高刻蚀温度或延长反应时间等实现Si的完全脱除.
图4是天然石墨原料和SiC@NG前驱体Cl2刻蚀后产物的Raman光谱,从图中可以看出,在约1330、1580和2670 cm-1处出现了三个峰,分别对应为石墨碳的D峰、G峰和2D峰.D峰归属于边缘或者其它缺陷形式存在的sp3杂化的碳原子,G峰对应于有序的sp2杂化的碳原子.从图中可看出,天然石墨呈现出强且半峰宽较窄的G峰和2D峰,表明其结构规整有序.25与天然石墨相比SiC-CDCs@NG的G峰明显变宽且对称性变差,而且呈现出强而宽的D峰,说明相当一部分sp2杂化碳原子转化为sp3杂化结构,即石墨晶体的有序度降低,产生了更多的缺陷,这主要是由于SiC@NG经氯气刻蚀所生成的衍生碳主要以无序的无定形碳为主.且随着NG/Si摩尔比的增大,D峰与G峰强度比(ID/IG)值逐渐减小,表明SiC-CDCs@NG结构中所含的无序的无定形衍生碳减少.这主要是因为NG/Si摩尔比越大,经固相反应生成的立方相SiC越少,刻蚀后得到的无定形衍生碳也越少.当NG/Si摩尔比为1时,SiCCDCs@NG的D峰强度超过G峰,且其2D峰已基本消失,说明此时结构中主要以无定形的衍生碳为主.此外,对于SiC-CDCs@NG-1,在785和961 cm-1出现的两个峰分别对应于SiC,它们分别对应于β-SiC的横光学声子模(TO)及纵光学声子模(LO)光学特征谱线.26,27表明材料中仍然残留部分没有刻蚀完全的SiC,这与图3 XRD的结果相一致.

图3 SiC-CDCs@NG-R的XRD图Fig.3 XRD patterns of SiC-CDCs@NG-R

图4 NG和SiC-CDCs@NG-R的Raman光谱Fig.4 Raman spectra of NG and SiC-CDCs@NG-R
图5是SiC@NG-2前躯体1000°C下经Cl2刻蚀制备的SiC-CDCs@NG的SEM图,复合材料仍然保持球形石墨的球形形貌,与刻蚀前的SiC@NG-2前躯体(图2(b,c))相比,球形石墨表面的粗糙度明显降低,颗粒明显减少,呈现较疏松的多孔状态.

图5 SiC-CDCs@NG-2的SEM图Fig.5 SEM images of SiC-CDCs@NG-2
图6是SiC@NG-2前驱体和SiC-CDCs@NG-2的TEM图,SiC@NG-2的TEM(图6a)及其HRTEM (图6b)中可以观察到明显的晶格条纹,其晶格条纹间距是0.251 nm,与立方β-SiC的(111)晶面族的晶面间距相等,由此可以进一步证实所得到的产物为立方的β-SiC单晶体.表明经固相反应后,在NG表面生成了新相β-SiC.经氯气刻蚀后,SiC-CDCs@NG-2的TEM图(图6c)及HRTEM图(图6d)中未观察到明显的SiC晶格条纹,主要以无序的无定形碳存在,其中有少量的短而卷曲的石墨烯层.表明经氯化反应后,SiC@NG-2中的Si元素基本被刻蚀掉,生成的碳化硅衍生碳有序性较低.从图6c可以清晰地观察到衍生碳包覆的核@壳结构,其壳的厚度大约在20 nm左右.

图6 (a,b)SiC@NG-2前驱体和(c,d)SiC-CDCs@NG-2的TEM图Fig.6 TEM images of(a,b)precursor SiC@NG-2 and(c,d)SiC-CDCs@NG-2
图7(a)是制备的SiC-CDCs@NG-R的N2吸/脱附等温线,从图中可以看出,所有的曲线都属于典型的I型曲线,即微孔的吸/脱附线,且随着NG/Si摩尔比的减小,在相对压力p/p0<0.1的区间内,N2的吸附量在不断增大,表明其微孔含量越高.图7(b)是根据DFT模型得到的SiC-CDCs@NG-R的孔径分布图,SiC-CDCs@NG-R的孔径可以分两类,即微孔(<2 nm)、中孔(2-50 nm).在SiC-CDCs@NG-R的孔径分布中以微孔为主,孔径主要集中在0.5-0.7 nm之间;随着NG/Si摩尔比减小,孔径分布更窄,微孔尺寸更小.
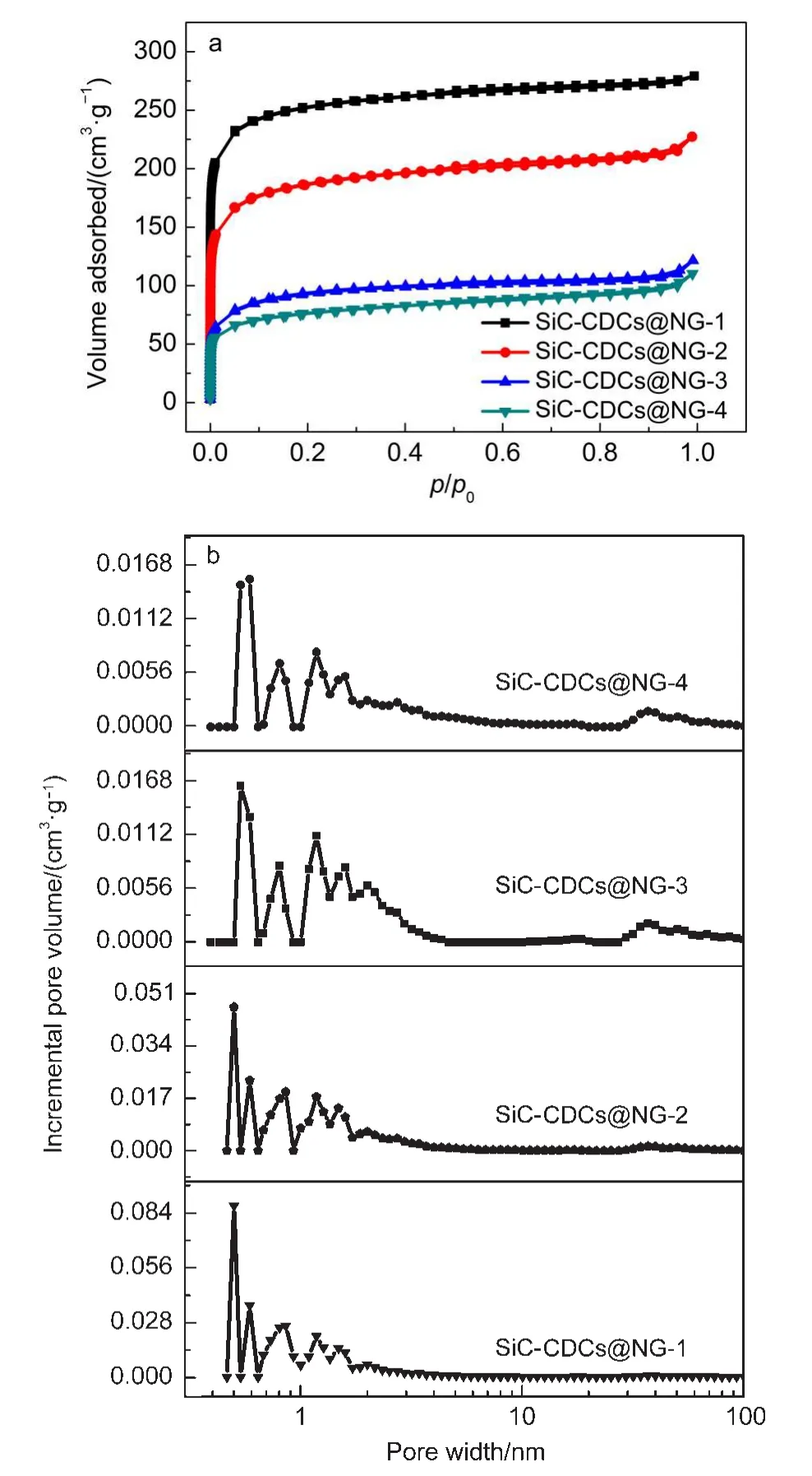
图7 SiC-CDCs@NG-R的(a)N2吸/脱附等温线和(b)孔径分布图Fig.7 (a)N2adsorption/desorption isotherms and(b) pore size distribution of SiC-CDCs@NG-R
表1列出了SiC@NG-1和SiC-CDCs@NG-R复合材料的结构参数,可以看出刻蚀前的SiC@CDC-1前驱体比表面积(1.8 m2·g-1)和总孔容(0.001 cm3· g-1)都很小,基本不含有微孔;而刻蚀后的产物孔体积(总孔体积和微孔体积)和比表面积都明显增大.随着NG/Si摩尔比减小,SiC-CDCs@NG-R的比表面积显著增大,由281.2 m2·g-1增加至964.1 m2·g-1,其比表面积主要由微孔贡献;材料总的孔体积和微孔孔体积也明显提高.
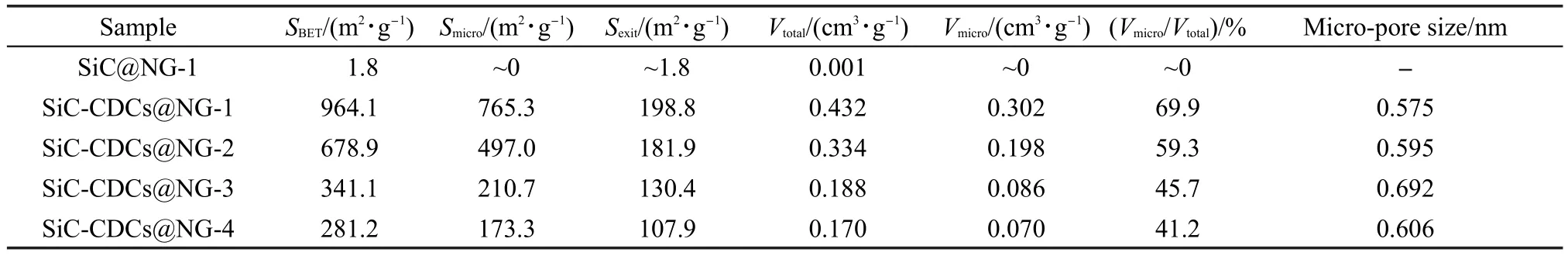
表1 SiC@NG-1和SiC-CDCs@NG-R复合材料的结构参数Table 1 Structure parameters of SiC@NG-1 and SiC-CDCs@NG-R composites
4 结论
采用固相法制备SiC@NG前驱体,然后通过氯气刻蚀的方法成功制备了SiC-CDCs@NG-R复合材料,该材料具有典型的核@壳结构,即以NG为核,以具有<1 nm的微孔的CDCs为壳.通过控制NG/Si摩尔比可以有效调控CDCs壳的厚度和NG核的尺寸,并在较大的范围内调节SiC-CDCs@NG-R的比表面积(281.2-964.1 m2·g-1)和孔体积(0.170-0.432 cm3·g-1).
(1)Xu,B.;Cao,G.P.New Carbon Mater.2008,23(1),95.[徐斌,曹高萍.新型炭材料,2008,23(1),95.]
(2)Zhang,R.J.;Zhou,B.Journal of Yanshan University2011,35 (4),283.[张瑞军,周 斌.燕山大学学报,2011,35(4),283.]
(3)Tsaia,W.Y.;Gao,P.C.;Daffos,B.;Taberna,P.L.;Perez,C.R.; Gogotsi,Y.;Favier,F.;Simon,P.Electrochem.Commun.2013,34,109.doi:10.1016/j.elecom.2013.05.031
(4)Lee,S.M.;Kaneko,K.Carbon2003,41(2),374.doi:10.1016/S0008-6223(02)00361-5
(5)Zhao,J.C.;Lai,C.Y.;Dai,Y.;Xie,J.Y.Mater.Lett.2007,61, 4639.doi:10.1016/j.matlet.2007.02.071
(6)Xu,J.;Wu,C.;Yan,P.T.;Zhang,R.J.;Yue,X.Q.;Ge,S.H. Microporous Mesoporous Mat.2014,198,74.doi:10.1016/j. micromeso.2014.07.019
(7)Becker,P.;Glenk,F.;Kormann,M.;Etzold,B.J.M.Chem. Eng.J.2010,159(1-3),236.doi:10.1016/j.cej.2010.02.011
(8)Ersoy,D.A.;McNallan,M.J.;Gogotsi,Y.G.Mater.Res.Innov.2001,5,55.doi:10.1007/s100190100136
(9)Zinovev,A.V.;Elam,J.W.;Moore,J.F.;Hryn,J.N.;Auciello, O.;Carlisle,J.A.;Pellin,M.J.Thin Solid Films2004,469-470, 135.
(10)Jia,J.;Yang,X.Y.;Yan,Y.;Zhu,Y.Y.;Xing,B.L.;Zhou,A.G. Chem.Ind.Eng.Prog.2014,33(10),2681.[贾 进,杨晓阳,闫 艳,朱元元,邢宝林,周爱国.化工进展,2014,33(10), 2681.]
(11)Urbonaite,S.;Wachtmeister,S.;Mirguet,C.;Coronel,E.;Zou, W.Y.;Csillag,S.;Svensson,G.Carbon2007,45(10),2047. doi:10.1016/j.carbon.2007.05.022
(12)Hoffman,E.N.;Yushin,G.;EL-Raghy,T.;Gogotsi,Y.; Barsoum,M.W.Microporous Mesoporous Mat.2008,112, 526.doi:10.1016/j.micromeso.2007.10.033
(13)Yachamaneni,S.;Yushin,G.;Yeon,S.H.;Gogotsi,Y.;Howell, C.;Sandeman,S.;Phillips,G.;Mikhalovsky,S.Biomaterials2010,31,4789.doi:10.1016/j.biomaterials.2010.02.054
(14)Leis,J.;Perkson,A.;Arulepp,M.;Kaarik,M.;Svensson,G. Carbon2001,39,2043.doi:10.1016/S0008-6223(01)00020-3
(15)Gogotsi,Y.;Welz,S.;Ersoy,D.A.;McNallan,M.J.Nature2001,411,283.doi:10.1038/35077031
(16)Seo,M.S.;Kim,J.H.;Kim,J.M.;Kang,S.;Ihm,J.S.;Kim,D. O.Carbon2013,60,299.doi:10.1016/j.carbon.2013.04.041
(17)Yeon,S.H.;Raddington,P.;Gogotsi,Y.G.;Fischer,J.E.; Vakifahmetoglu,C.;Colombo,P.Carbon2010,48,201.doi: 10.1016/j.carbon.2009.09.004
(18)Kormann,M.;Gerhard,H.;Popovska,N.Carbon2009,47, 242.doi:10.1016/j.carbon.2008.10.002
(19)Welz,S.;McNallana,M.J.;Gogotsi,Y.J.Mater.Process. Technol.2006,179,11.doi:10.1016/j.jmatprotec.2006.03.103
(20)Sun,X.Z.;Zhang,X.;Huang,B.;Ma,Y.W.Acta Phys.-Chim. Sin.2014,30,485.[孙现众,张 熊,黄 博,马衍伟.物理化学学报,2014,30,485.]doi:10.3866/PKU.WHXB201401131
(21)Zhai,X.L.;Song,Y.;Li,P.;Guo,Q.G.;Zhi,L.J.Chemical Research2014,25(2),112. [翟晓玲,宋 燕,李 鹏,郭全贵,智林杰.化学研究,2014,25(2),112.]
(22)Zhong,H.X.;Zhao,C.B.;Luo,H.;Zhang,L.Z.Acta Phys.-Chim.Sin.2012,28,2641.[仲皓想,赵春宝,骆 浩,张灵志.物理化学学报,2012,28,2641.]doi:10.3866/PKU. WHXB201207181
(23)Liu,G.Y.;Zhou,A.N.Carbon2006,4,27.[刘国阳,周安宁.炭素,2006,4,27.]
(24)Sun,G.;Song,W.;Liu,X.;Qiao,W.;Long,D.;Ling,L.Mater. Lett.2011,65,1392.doi:10.1016/j.matlet.2011.02.011
(25)Jeong,M.G.;Yoon,S.H.;Chun,Y.S.;Lee,E.S.;Lim,D.S. Carbon2014,79,19.doi:10.1016/j.carbon.2014.07.016
(26)Sui,J.;Liu,X.F.Surf.Coat Technol.2013,235,469.
(27)Liu,X.F.;Huang,Q.Z.;Su,Z.A.;Jiang,J.X.J.Chin.Ceram. Soc.2004,32(7),906.[刘兴防,黄启忠,苏哲安,蒋建献.硅酸盐学报,2004,32(7),906.]
Preparation and Structure Regulation of Silicon Carbide-Derived Carbon/ Spherical Natural Graphite Composites
DU Xue-Lian1,2CONG Ye1,2,*JIANG Lu2LI Xuan-Ke1,2,*CUI Zheng-Wei2DONG Zhi-Jun2YUAN Guan-Ming2ZHANG Jiang2
(1The State Key Laboratory of Refractories and Metallurgy,Wuhan University of Science and Technology,Wuhan 430081, P.R.China;2Hubei Province Key Laboratory of Coal Conversion&New Carbon Materials,Wuhan University of Science and Technology,Wuhan 430081,P.R.China)
To meet the requirements of the energy storage materials for high energy density and high power density,porous silicon carbide/derived carbon-spherical natural graphite(SiC-CDCs@NG)composites were prepared.The composites were composed of tailored porous carbide-derived carbon and graphitized carbon with excellent conductivity and a high energy storage capacity.The composition,structure,morphology,pore structure,and specific surface area of the composites were characterized by X-ray diffraction(XRD),scanning electron microscopy(SEM),transmission electron microscopy(TEM),Raman spectroscopy,and N2adsorption/ desorption analysis.The composites exhibited a high and adjustable specific surface area and micro-pore volume,with a micro-pore size of between 0.5 and 0.7 nm.Varying the molar ratio of NG and Si allowed optimization of the micro-pore volume,pore size distribution,specific surface area,and composition and content of the CDCs shell and NG core.
Silicon carbide-derived carbon;Composite;Structure regulation;Specific surface area; Pore size distribution
O648;TQ127.1
10.3866/PKU.WHXB201501212www.whxb.pku.edu.cn
Received:December 9,2014;Revised:January 20,2015;Published on Web:January 21,2015.
∗Corresponding authors.CONG Ye,Email:congye626@126.com.LI Xuan-Ke,Email:xkli8524@sina.com;Tel:+86-27-86556906. The project was supported by the National Natural Science Foundation of China(51472186,51402221).
国家自然科学基金(51472186,51402221)资助项目
