分散液液微萃取-分光光度法测定水中痕量铜
2015-12-24徐鉴邵阳张翠玲
徐鉴,邵阳,张翠玲
(南京晓庄学院 生物化工与环境工程学院,江苏 南京 211171)
微量元素铜是动植物和人体内的主要微量营养元素之一[1],参与了体内多种代谢。但如果摄取量过多就会引发多种疾病[2]。随着工业化的日益发展,环境中的铜污染变得更加严重。因此,研究准确测定环境中痕量铜的方法具有重要意义。目前测定铜的分析方法主要有分光光度法[3]、原子吸收光谱法[4]、电感耦合等离子体-原子发射光谱法[5]、电化学法[6]等。Assadi 等[7]报道了分散液液微萃取(DLLME),该方法集采样、萃取和浓缩于一体,操作简便、快速、成本低、对环境污染小且富集效率高。
1 实验部分
1.1 试剂与仪器
Cu(NO3)2·3H2O,优级纯;盐酸羟胺、新铜试剂(2,9-二甲基-1,10-菲罗啉)、甲醇、醋酸-醋酸钠缓冲溶液均为分析纯;实验用水为二次蒸馏水。
UV2450 型紫外可见分光光度计;AUY120 型电子天平;CT14D 型高速离心机;PHS-3C 型精密pH计。
1.2 溶液制备
1.2.1 1.0 mg/mL 铜(Ⅱ)标准贮存溶液 准确称取优级纯Cu(NO3)2·3H2O 3.801 9 g 溶于水,移入1 000 mL 容量瓶中,摇匀,以水定容。
1.2.2 0. 1 mmol/mL 盐酸羟胺溶液 准确称取0.694 9 g 盐酸羟胺溶于100 mL 水中。
1.2.3 0.1 mmol/mL 新铜试剂溶液(2,9-二甲基-1,10-菲罗啉)(螯合剂) 准确称取2.082 6 g 新铜试剂溶于100 mL 甲醇中。
1.3 实验方法
准确移取5 mL 浓度为100 μg/L 的Cu2+溶液于20 mL 的具塞离心管中,加入缓冲溶液调节溶液pH=6,再加入1 mL 的0.1 mmol/mL 盐酸羟胺溶液,反应5 min 后,此时溶液中的Cu2+被还原成Cu+。此时再向溶液中加入1 mL 的0.1 mmol/mL新铜试剂溶液,摇匀,静止3 min。加入1 mL 的乙腈后用微量进样器快速注入100 μL 氯仿,剧烈振荡2 min,得到均匀的乳浊液,3 000 r/min 离心3 min后,将有机相沉淀全部转移到刻度离心管中,加入甲醇,稀释至5 mL,混匀后,置于石英比色皿中,以空白试剂为参比,在430 nm 处测定其吸光度。
2 结果与讨论
2.1 吸收光谱
按实验方法,在300 ~600 nm 范围内扫描溶液的吸收光谱,见图1。
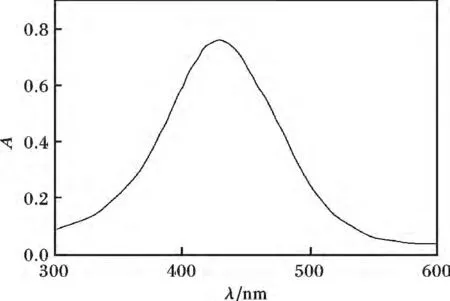
图1 铜-新铜试剂配合物的吸收光谱Fig.1 Absorption spectra of Cu-neocuproine complex
由图1 可知,铜络合物最大吸收峰位于430 nm处,试剂空白不干扰测定,故选择430 nm 为测定波长。
2.2 萃取剂的选择
萃取剂的选择是影响DLLME 萃取效率的一个关键因素。萃取溶剂必须满足以下3 个条件:①具有比水更大的密度,以便通过离心的方式把水溶液与萃取溶剂分离;②对待测物具有比水更大的溶解度,以保证取得良好的萃取效率;③当萃取剂注入水溶液时,能形成含有分散剂的微小液滴混浊溶液。按照实验方法考察了氯仿、四氯化碳、氯苯、溴苯等溶剂对萃取效率的影响,实验结果表明,氯仿的萃取效率最高。因此,本实验选择氯仿作为萃取剂。
2.3 萃取剂的用量
在保证萃取完全的前提下,尽可能减少相体积比,可以提高萃取效率和富集能力。按照实验方法考察了氯仿体积从50 μL 到150 μL(间隔10 μL)对吸光度的影响。实验结果表明,当氯仿的体积为100 μL 时,吸光度达到最大且保持稳定,故最终选择萃取剂的体积为100 μL。
2.4 分散剂的选择
分散剂的选择是影响DLLME 萃取效率的另一个关键因素。要求分散剂不仅在萃取溶剂中有良好的溶解性而且还能与水互溶,使萃取溶剂在水相中分散成细小的液滴,增大其与待测物的接触面积,从而提高萃取效率。按照实验方法,考察了甲醇、乙醇、丙酮、乙腈等分散剂,当选择甲醇和乙醇作为分散剂时对氯仿在水中不能形成较为稳定的乳浊液,而丙酮和乙腈则可以形成稳定的乳浊液,其中以乙腈为分散剂时所形成的乳浊液更为明显,富集倍数更高。因此,本实验选择乙腈为分散剂。
2.5 分散剂的用量
按照实验方法考察了分散剂乙腈体积从0.5 mL 到1.5 mL(间隔0.1 mL)对吸光度的影响。实验结果表明,当乙腈的体积为1.0 mL 时,吸光度最大且保持稳定。这是因为乙腈用量小时,萃取剂不能很好的分散在水相中,导致萃取效率低;当乙腈用量较大时,被测组分在水中的溶解度增大,分析物不易被萃取剂富集而使萃取效率降低。故最终选择分散剂体积为1.0 mL。
2.6 螯合剂的用量
螯合剂的主要作用是与金属离子形成稳定的疏水性配合物,使金属离子萃取到有机相中。按照实验方法考察了浓度为0.1 mmol/mL 新铜试剂体积从0.5 mL 到1.5 mL(间隔0.1 mL)对吸光度的影响。实验结果表明,当新铜试剂的体积为1.0 mL时,吸光度达到最大且保持稳定,故最终选择浓度为0.1 mmol/mL 新铜试剂体积为1.0 mL。
2.7 pH 值及缓冲溶液用量的影响
溶液的pH 值影响疏水性配合物的形成,从而影响萃取效率。按照实验方法,考察了pH 在4 ~8范围内吸光度的变化情况。实验结果表明,当pH=6 时吸光度可达最大值且稳定。本实验选用pH =6醋酸-醋酸钠缓冲溶液。
实验了不同体积pH =6 的醋酸-醋酸钠缓冲溶液对Cu2+萃取的影响。实验结果表明,pH =6 的醋酸-醋酸钠缓冲溶液的最佳用量为0.5 ~1. 0 mL。本实验选用醋酸-醋酸钠缓冲溶液1.0 mL。
2.8 盐浓度的影响
按照实验方法考察了离子强度对萃取效率的影响。在水相中加入不同浓度的NaCl 溶液(0 ~5%)。实验结果表明,随着盐浓度的增加,体系的吸光度无明显变化。故本实验选择不加盐。
2.9 萃取时间的影响
在DLLME 过程中,萃取时间是指在水相中加入分散剂和萃取剂后到开始离心之前的这段时间间隔。按照实验方法考察了1 ~5 min 时间范围内对萃取效率的影响。实验结果表明,萃取时间为2 min时吸光度可达最大值,所需萃取时间很短,这是因为溶液形成乳浊液之后萃取溶剂被均匀的分散在水相中,与待测物接触面积大,待测物由水相转移到有机相并且迅速地达到两相平衡。故本实验选择萃取时间为2 min。
2.10 共存离子的影响
按实验方法考察了实际样品中可能存在的离子对铜离子测定的影响。对100 μg/L 的Cu2+标准溶液进行测定,当相对误差不超过±5%时,下列共存物质不干扰测定:Li+、Na+、K+(100 mg/L);Ca2+、Mg2+、Pb2+、Sn2+、Mn2+(70 mg/L);Fe3+、Ag+、Ba2+(50 mg/L);Zn2+、Co2+、Ni2+、Cd2+(20 mg/L)。可见体系对铜离子检测具有很高的选择性。
2.11 工作曲线、检出限和精密度
配制一系列铜离子标准溶液,按实验方法,在430 nm 处测定其吸光度。结果表明,铜离子浓度在1.0 ~200 μg/L 范围内与吸光度呈良好的线性关系,线 性 回 归 方程 为A = 0. 003 2 + 0. 043 6C(μg/L),相关系数r =0.998 4。由11 次空白测定的标准偏差的3 倍除以工作曲线的斜率,求得本方法检出限为0.35 μg/L。对含有100 μg/L 的铜离子标准溶液平行萃取测定11 次,测定结果的RSD 为3.42%。
2.12 样品分析
按实验方法检测了自来水、河水和工业废水中的铜离子。对样品平行测定5 次,样品的加标回收率在96.2% ~101.7%,结果见表1。

表1 实际水样的测定结果及加标回收率Table 1 The determination results and spike recovery of real water
3 结论
本实验建立了以氯仿为萃取剂,乙腈为分散剂,新铜试剂为螯合剂的分散液液微萃取方法,将该方法与发光光度法联用测定水中的痕量铜。该方法集采样、萃取和浓缩于一体,具有操作简单、快速、灵敏度高等优点,适用于各种实际水样中痕量铜的测定。
[1] Kramer R.Fluorescent chemosensors for Cu2+ions:Fast,selective,and highly sensitive[J]. Angew Chem Int Ed,1998,37(6):772-773.
[2] 王秋莲,翁红华.儿童保健门诊524 例儿童微量元素检测结果分析[J]. 临床和实验医学杂志,2006,5(12):1970-1973.
[3] 凌育赵.吐温-80-DDTC 胶束增溶吸光光度法测定饮料中微量铜[J].理化检验:化学分册,2006,42(7):536-538.
[4] 刘汉东,刘琼玉,胡德文,等.流动注射在线液-液萃取FAAS 法测定地表水中痕量铜[J].光谱实验室,2000,17(6):690-693.
[5] 王艳君,蒋晓光,李卫刚,等.ICP-AES 法测定红土镍矿中镍、钙、钛、锰、铜、钴、铬、锌与磷的含量[J].分析试验室,2012,31(9):50-53.
[6] 朱永春,李丹,岳爽,等.超声波-微分脉冲伏安法测定超痕量铜[J].分析化学,2006,34(5):721-724.
[7] Rezaee Mohammad,Assadi Yaghoub,Hosseini Mohammad-Reza Milani,et al.Determination of organic compounds in water using dispersive liquid-liquid microext raction[J].Journal of Chromatography A,2006,1116(122):1-9.
