复方白芍颗粒质量控制研究
2015-12-19储晓琴桂双英
王 帆,储晓琴,桂双英
(1.安徽中医药大学药学院,安徽 合肥 230012;2.安徽中医药高等专科学校,安徽 芜湖 241000;3.安徽省中药制剂工程技术研究中心,安徽 合肥 230012;4.安徽省中医药科学院药物制剂研究所,安徽 合肥 230012)
复方白芍颗粒由白芍、甘草、泽泻等中药组成,为安徽省六安地区民间祖传经验方,具有滋阴健肾、活血化瘀、除湿化浊的功效。原方采用药材打粉直接用水冲服给药,临床用于治疗慢性肾炎、肾功能不全,效果显著。本课题组采用现代提取与制剂技术研制了复方白芍颗粒[1],为了更好地控制制剂质量,本实验对其质量标准进行研究。白芍和泽泻分别是该方中的君药和臣药,芍药苷为治疗慢性肾病的主要活性物质,白芍和泽泻的薄层鉴别方法及芍药苷的含量测定方法比较成熟稳定,因此本实验以白芍和泽泻作为鉴别考察的依据,芍药苷作为定量指标来控制复方白芍颗粒质量。
1 仪器与试药
1.1 仪器 LC-20A高效液相色谱仪:日本岛津公司(配有SPD-M20紫外检测器,LC-20AB泵,LC solution色谱工作站);AG285型电子天平:德国METTLER公司;循环水式多用真空泵、R-1001N型旋转蒸发仪:河南省郑州长城科工贸有限公司。
1.2 试药 芍药苷对照品(批号110736-201035):中国药品生物制品检定所;23-乙酰泽泻醇B对照品(批号 MUST-11040101):四川省成都曼思特生物科技有限公司;薄层层析用硅胶G、薄层层析用硅胶H:山东省青岛海洋化工厂;复方白芍颗粒为自制(批号分别为121024、121120、121209);阴性对照品为自制;甲醇、乙腈均为色谱纯;水为娃哈哈纯净水;其余化学试剂均为分析纯。
2 方法与结果
2.1 白芍的鉴别[2-5]取本品颗粒约2.0g,研成细粉,加甲醇10mL,超声波(功率100W,频率40 kHz)30min,滤过,滤液蒸干,残渣加入1mL甲醇使其溶解,即得供试品溶液;另外取适量的芍药苷对照品,加入甲醇适量,使成1mg/mL的溶液,即得对照溶液;取缺白芍提取物的阴性样品,按照上述方法制备阴性对照液;按照《中华人民共和国药典》2010年版一部附录ⅥB薄层色谱法(thin layer chromatography,TLC)试验要求,精密吸取上述3种溶液各10μL,分别点在同一硅胶G薄层色谱板上,将三氯甲烷-甲醇-乙酸乙酯-甲酸(40∶10∶5∶0.2)作为展开剂,展开,晾干,喷洒显色剂5%的香草醛硫酸溶液适量,加热直至斑点颜色显示清晰。在供试品色谱图中,与对照品色谱图的对应位置上,显示相同颜色的蓝紫色斑点,阴性对照溶液无干扰。结果见图1。
2.2 泽泻的鉴别[2]213取本品颗粒约2.0g,研成细粉,加入乙酸乙酯20mL,超声波处理30min,滤过,将滤液加于氧化铝柱(200~300目,5g,内径1cm,干法装柱)上,用乙酸乙酯10mL洗脱,收集洗脱液,蒸干,残渣加入1mL乙酸乙酯使其溶解,作为供试品溶液。取23-乙酰泽泻醇B对照品适量,加乙酸乙酯制成2mg/mL的溶液作为对照品溶液。取缺泽泻提取物的阴性样品同法制成阴性对照溶液;按照TLC法(《中华人民共和国药典》2010年版一部ⅥB)试验,精密吸取上述3种溶液各10μL,分别点在同一硅胶H薄层板上,以乙酸乙酯-环己烷(1∶1)为展开剂,展开,取出,晾干,喷洒显色剂5%的硅钨酸乙醇溶液适量,在105℃下加热至斑点颜色显示清晰。供试品色谱图中,在与对照品色谱图相应位置上,显示相同颜色的红色斑点,阴性对照溶液无干扰。结果见图2。
2.3 芍药苷含量测定[6-10]
2.3.1 色谱条件 色谱柱为phenomenex C18(250 mm×4.6mm,5μm);流动相:乙腈-0.05%磷酸水溶液(14∶86);流速:1.0mL/min;检测波长:230nm;柱温:30℃;进样量:20μL。理论塔板数按照芍药苷的峰计算应高于2 000,分离度应不低于1.5。
2.3.2 对照品溶液制备 取经干燥的芍药苷对照品适量,精密称取33.35mg,置于50mL量瓶中,加甲醇稀释至刻度,制成0.667mg/mL的芍药苷对照品贮备溶液。吸取对照品贮备溶液1mL置10mL量瓶中,加入甲醇至刻度,即得66.7μg/mL的芍药苷对照品溶液。
2.3.3 供试品溶液的制备 取本品适量,研细,精密称定0.5g,置50mL量瓶中,加甲醇至刻度,超声提取30min,放冷,用甲醇补足缺失量,用0.45 μm微孔滤膜滤过,取续滤液,即得。
2.3.4 阴性对照溶液 按制剂工艺制备处方中除去白芍的阴性对照样品,照“2.3.3”项下方法制备阴性对照溶液。分别精密吸取对照品溶液、供试品溶液、阴性对照溶液各20μL,按含量测定项下方法测定,色谱图(见图3)提示,在对照品色谱相应的位置上,供试品溶液具有相同保留时间的色谱峰,阴性对照在此峰位无吸收。
2.3.5 线性关系考察 分别精密量取芍药苷对照品贮备溶液(0.667mg/mL)0.1、0.2、0.4、0.6、0.8、1.0mL,分别置2mL量瓶中,用甲醇稀释至刻度,摇匀,过0.45μm微孔滤膜,制成系列对照品溶液。按“2.3.1”项下方法测定,以峰面积y对芍药苷浓度x(μg/mL)进行线性回归,即得回归方程:y=29 505x-108 325,r=0.999 1(n=6)。结果表明,芍药苷浓度在33.35~333.50μg/mL范围内与峰面积的线性关系良好。
2.3.6 精密度考察 精密量取芍药苷对照品溶液20μL,连续进样6次,测得芍药苷的峰面积,结果表明此方法的精密度良好(RSD=1.20%)。
2.3.7 重复性考察 取相同批号样品6份,按照“2.3.3”项下的方法制备样品溶液,测得芍药苷的峰面积,计算其平均含量为2.30%,RSD为1.98%(n=6),表明该方法重复性良好。
2.3.8 稳定性考察 取同一样品溶液,分别于0、1、2、4、8、12h进样,测定芍药苷峰面积,结果其RSD为2.0%,表明样品溶液在12h内基本稳定。
2.3.9 加样回收率考察 称取6份已知含量的样品(批号121120)约0.1g,精密称定,置50mL量瓶中,分别精密加入浓度0.667mg/mL对照品贮备溶液4mL,按照“2.3.3”项下方法操作,测定芍药苷含量,结果见表1。

表1 加样回收率试验结果
2.3.10 样品含量测定 按“2.3.3”项下方法制备供试品溶液(批号分别为121024、121120、121209),分别吸取对照品溶液和供试品溶液各20μL,注入高效液相色谱仪,按色谱条件测定峰面积,以外标一点法计算芍药苷含量。结果见表2。
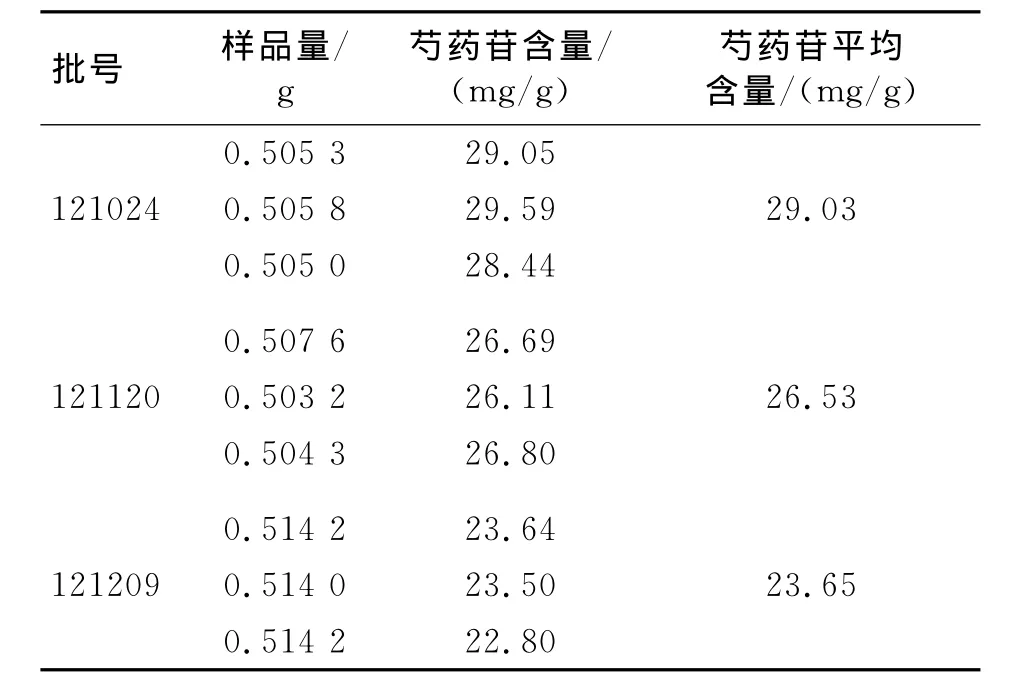
表2 样品含量测定结果
3 讨论
3.1 超声时间的确定 在含量测定时,采用超声提取法制备供试品溶液,对提取溶剂和超声时间进行考察,分别选择甲醇、50%乙醇、70%乙醇、无水乙醇超声30min,结果甲醇明显优于不同浓度乙醇;再分别以甲醇超声提取20、30、40min,结果甲醇超声提取30min与40min提取率相近,并高于20min,故选择甲醇超声提取30min。
3.2 流动相的选择 曾采用乙腈-水(14∶86)为流动相,但存在明显的拖尾现象,选用乙腈-0.05%磷酸(14∶86)作流动相时,可改善拖尾现象,且峰型较好。因此,本实验选取乙腈-0.05%磷酸溶液(14∶86)为流动相。
3.3 线性关系考察 芍药苷浓度在33.35~333.5 μg/mL范围内与峰面积的线性关系良好(r=0.999 1),不仅说明方法可靠,而且说明无论是用标准曲线还是用外标一点法计算含量,其结果区别不大,因此,在样品测定中采用外标一点法计算芍药苷含量,简单准确。
[1]王帆,桂双英,王举涛.正交试验法优选复方白芍颗粒提取工艺[J].安徽中医学院学报,2012,31(6):71-73.
[2]国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2010:97,213.
[3]程巧鸳,陈碧莲.明目地黄丸(浓缩丸)质量标准研究[J].中成药,2012,34(6):1091-1095.
[4]任爱农,孔铭,田耀洲.清清颗粒的质量标准研究[J].中成药,2007,29(3):456-458.
[5]王亚洲,杨春旭.蝎龙酒质量标准研究[J].中国实验方剂学杂志,2012,18(23):105-108.
[6]李越峰,杨武亮,沈菲,等.高效液相色谱测定白芍中芍药苷的含量[J].时珍国医国药,2008,19(2):438-439.
[7]梁秀琼,钟镜金,李卓明,等.调经丸质量标准研究[J].中药新药与临床药理,2007,18(5):388-390.
[8]施钧瀚,牛晓静,孙明明.慢肝康丸的质量标准研究[J].中国实验方剂学杂志,2010,16(16):42-44.
[9]郭炜,袁志芳,张兰桐.归芍软胶囊质量标准研究[J].中成药,2006,28(3):351-355.
[10]罗建明,李智勇.归附调经颗粒的质量标准研究[J].中药材,2011,34(8):1295-1298.
