血管外膜成纤维细胞及其还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶激活在血管损伤中的作用*
2015-12-15王洋综述雷燕审校
王洋综述,雷燕审校
血管外膜成纤维细胞及其还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶激活在血管损伤中的作用*
王洋综述,雷燕审校
血管外膜是血管壁最复杂的组成部分,血管外膜成纤维细胞(AF)是血管外膜主要的细胞成分,在血管的病理生理过程中发挥着重要作用。AF的还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶激活与活性氧生成是多种血管损伤的始发阶段和共同机制。本综述对AF及其NADPH氧化酶、活性氧在血管损伤中的作用进行了总结。
血管外膜成纤维细胞;还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶;血管损伤
血管壁是一个有序的层状结构。肌性和弹性的大中动脉(主动脉和冠状动脉等),其管壁从管腔面向外一般依次分为内膜、中膜和外膜。在血管损伤的研究历程中,外膜的作用一度为人们忽视:20世纪70年代之前,血管疾病的研究主要聚焦于中膜;20世纪70年代末,内皮细胞舒血管因子的发现掀起了学界研究内膜的热潮;20世纪90年代以来,研究者逐渐发现,外膜具有神经内分泌等多种重要功能,在血管生理病理进程中发挥着举足轻重的作用。外膜不再是血管疾病的“旁观者”,这个血管生物学中的新前沿,引起了各方向研究者关注。
1 外膜是结构最复杂的血管壁成分
与单纯由内皮细胞组成的内膜和以平滑肌细胞为主的中膜相比,血管外膜的成分相对复杂。首先,外膜包含丰富的细胞外基质,其主要成分是弹性纤维和胶原蛋白,其中分布着细胞、神经末梢、滋养血管、淋巴组织等多种成分;除此之外,外膜中还分布着种类繁多的细胞,包括成纤维细胞、脂肪细胞、免疫细胞、祖细胞、干细胞等。细胞间通过直接接触、自分泌或旁分泌的作用彼此交流并与相邻组织细胞相互感应,具有感知刺激并直接作出响应的能力。越来越多的文献支持这一观点:外膜是血管壁主要的“损伤感应器”,在激素、炎症或缺氧、缺血等刺激下,外膜细胞首先反应,发生细胞增殖、收缩表达、干细胞激活、黏附因子上调,分泌炎症因子、生长因子、血管生成因子等过程,进而导致管壁功能和形态的改变。
2 血管外膜成纤维细胞是血管壁的“哨兵细胞”
血管外膜最主要的细胞成分是血管外膜成纤维细胞(AF)。AF具有多源性,至少包括三种细胞来源:原始间充质、上皮间质、骨髓母细胞。此外,AF具有器官和组织特异性,这一点与白细胞类似,如纤维生发环境中的成纤维细胞(如肺AF),较之于普通组织的成纤维细胞,对增生刺激更为敏感。存在于相同器官/组织的AF以多种干细胞亚型的形式存在,一旦感受缺氧或其他因素的刺激,每一种亚型都可能做出特异反应,并行使其相应的功能。AF在一系列血管病理生理活动中,发挥着不可替代的作用。一方面,AF分泌胶原蛋白形成血管骨架,产生细胞因子等活性物质促进管壁其他细胞生长,参与血管形成;另一方面,当血管受损时,AF激活并发生表型改变,进而增殖,迁移,分泌胶原蛋白,参与血管修复。AF在血管应激及损伤修复中发挥着极其重要的作用,在血管受到刺激时,AF最先作出响应并活化,是血管壁的“哨兵细胞”,内皮细胞和平滑肌细胞都不能与之相提并论[1]。
AF在血管生长发育中的作用:血管系统的形成是一个极其复杂的过程,医学界对此尚无完整认识。可以肯定的是,此过程既受基因调控的影响,也被局部环境因素所刺激:基因决定了血管生长的主要形式,缺氧、营养不足等局部因素影响血管的生长。研究发现,血管外膜有类似干细胞和祖细胞巢的作用,外膜中分离的干细胞具有发育成内皮细胞、壁细胞、成骨细胞及脂肪细胞等多种细胞的能力,说明外膜在血管壁生长、重塑和修复中具有扮演了重要的角色[2],而AF和还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶系统则促成了血管和原始内皮血管的生成[3]。
AF在血管中膜增生过程中的作用:AF的NADPH氧化酶-活性氧系统参与了血管中膜的增生。在血管紧张素II(AngII)促中膜增生的实验中,人们发现AngII在导致中膜增生的同时激发了小鼠主动脉外膜的NADPH氧化酶-活性氧系统,对外膜NADPH氧化酶水平偏低的nox2缺乏小鼠,AngII的刺激大大降低,说明外膜的NADPH氧化酶-活性氧系统对中膜具有旁分泌作用[4]。用搭载NADPH氧化酶p47phox阻断剂的腺病毒干扰大鼠颈动脉AF,结果显示,阻断剂能显著降低中膜活性氧的生成和中膜肥厚,进一步证实了AF的NADPH氧化酶-活性氧系统促进中膜增生的重要作用[5]。
AF在血管内膜生长过程中的作用:AF的增生、迁移是血管内膜生长、重塑的病理基础。Scott等[6]对小型家猪实施了左冠状动脉前降支和回旋支的经皮腔内冠状动脉成形术,手术后的初始时间点即发现增殖细胞出现在血管外膜,除血管撕裂处细胞增生旺盛外,整个脉道都有细胞增生,更加有趣的是,直至手术7天后,外膜的细胞增殖才趋于缓和,而此时,内膜细胞增殖才逐步旺盛。Wilcox等[7]将血管外膜称为冠状动脉成形术后“细胞增殖的先锋部队”。Li等[8]的工作支持了Scott的发现,他们的实验检测到气囊损伤后血管外膜肌成纤维细胞迁移到内膜,进一步证实了AF对内膜生长的贡献。
AF与细胞外基质:血管外膜的细胞外基质主要由胶原构成。胶原的组成和含量决定了血管壁弹性和韧性,其变化在高血压、动脉粥样硬化、血管重塑等病变中具有重要意义。外膜的胶原成分主要为Ⅰ、Ⅲ型胶原,主要由AF分泌,AF决定了血管外膜细胞外基质的组成。一般情况下,AF处于休眠状态,即未分化类型,在压力或损伤条件下,AF激活并行使分泌功能,细胞外基质即发生量和质的改变,从而进一步导致血管结构和功能的变化。在血管再狭窄、粥样硬化和肺动脉高压等血管疾病的进程中,血管外膜的基质成分发生了明显的改变。研究发现,自发性高血压大鼠(SHR)AF的Ⅰ、Ⅲ型胶原免疫细胞染色阳性,胸主动脉胶原沉积高于对照Wistar大鼠,说明AF可以合成Ⅰ、Ⅲ型胶原,并通过此途径参与了血管损伤[9]。
AF向肌成纤维细胞的转化:肌成纤维细胞指具有某些肌纤维细胞特点的成纤维样细胞。Gabbiani等[10]首先报道了成纤维细胞向肌成纤维细胞的转化过程:在损伤修复或纤维化过程中,活化的成纤维细胞获得了平滑肌细胞的特征,包括结蛋白、钙介素、平滑肌肌球蛋白重链和肌动蛋白的一个亚型(α-SMA) 。肌成纤维细胞具有多源性,除成纤维细胞外,上皮细胞、内皮细胞和干细胞均能分化成为肌成纤维细胞。肌成纤维细胞最常见的标记物是α-SMA,但其不能甄别肌成纤维细胞的来源,也不能区别肌成纤维细胞与平滑肌细胞[11]。目前尚未发现肌成纤维细胞的特异性蛋白。
肌成纤维细胞可以生成胶原蛋白和其他细胞外基质、生长因子、细胞因子和活性氧等,并对中膜平滑肌细胞具有旁分泌作用,进而导致血管重塑等变化,是组织重塑的关键参与者[12]。如动静脉瘘成形术后,静脉段血管外膜肌成纤维细胞激活,促使中膜增厚和外膜纤维化,最终导致动静脉瘘堵塞[13]。由此可见,AF向肌成纤维细胞的转化可能是血管损伤应答的关键一环。
3 血管外膜还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶是血管壁氧化产物的主要生产者
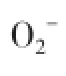
活性氧家族,包括超氧化物、过氧化氢和过氧亚硝酸盐等。哺乳动物细胞生成活性氧,活性氧对大分子物质如核糖核酸(RNA)、脱氧核糖核酸(DNA)、蛋白质和脂类产生氧化和硝化作用,从而损伤细胞。与此同时,活性氧通过促进生长因子和转录因子对细胞信号通路的调节,促进细胞的增殖、分化和凋亡。氧化应激出现在多种疾病中,这一过程不仅是活性氧生成和清除的失衡,更是产生活性氧的各种酶的功能紊乱。


NADPH氧化酶在氧化应激中的地位如此重要,因此,人们试图减低其活性甚至抑制其作用。如细胞实验发现,坎地沙坦可通过下调 NADPH氧化酶p22phox亚基的表达,减少血管外膜成纤维细胞内活性氧的产生[18]。NADPH氧化酶衍生的氧化应激反应的负向调节因子——小窝蛋白-1(caveolin-1)也被发现,它主要分布于血管内皮和外膜,其表达的缺失可以提高nox的活性,促进活性氧的生成,与心血管疾病相关[19]。还有研究发现,NADPH氧化酶启动的氧化应激作用可以被E1激活基因遏制子(CREG)所抑制,这一作用可能与其直接抑制成纤维细胞NADPH蛋白酶产生活性氧,间接抑制NADPH氧化酶下游的细胞外信号调节激酶(ERK1/2), c-Jun氨基酸末端激酶(JNK) 和p38丝裂原活化蛋白激酶(p38 MAPK)的表达有关[20]。
4 血管外膜成纤维细胞还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶激活是血管损伤的导火索

动脉粥样硬化(AS):动脉粥样硬化病变始于外膜,AF活化是AS的始发因素。对人类的病理学研究证明了这一点:有学者对108名平均年龄为34.6岁的肯尼亚黑种人进行尸检,结果发现,14.8%的左前降支和11.1%的颈动脉外膜出现了增厚,即动脉粥样硬化的特点之一,其中,6.5%的左前降支和3.7%的外膜增厚不伴随内膜增生,说明外膜的病变发生在中膜和内膜受损之前[24]。AS早期就存在AF活化,活化的AF合成活性氧、一氧化氮(NO)、炎性因子、活性肽等多种活性因子,在内膜损伤发生前就启动了AS的发生。将表达β-gal的腺病毒载体标记在大鼠颈动脉内膜拉伤模型的AF上,结果发现,动脉损伤后AF由外膜迁移到内膜,为AF参与AS提供了直接证据[25]。另有研究指出,Ang II可通过酪氨酸激酶介导的信号转导途径激活NADPH氧化酶,增加AF的活性氧生成,进而促进血管炎性反应和AS的发展[26]。不仅如此,AF在NADPH氧化酶作用下产生内源性O2-等活性氧,导致氧化型低密度脂蛋白的生成增加,从而进一步加剧AS[27]。
血管重塑 :AF是血管重塑过程中的重要角色。高血压、动脉粥样硬化和动脉损伤等刺激均能活化AF,活化的AF不仅增殖、迁移、参与中膜和内膜的形成,而且通过分泌胶原蛋白进而导致血管收缩和纤维化,后者恰恰是病理性血管重塑的特征性改变。在血管损伤的早期,AF发挥着“第一响应者”的作用。如Arribas等[28]在慢性一氧化氮抑制收缩压升高模型上发现,3周高血压刺激后,外膜增厚,而中膜和内膜没有增厚,呈鲜明对比。AF到肌成纤维细胞的表型改变是血管重塑的基本特点,实验发现,AngII介导了AF向肌成纤维细胞的转变,其具体途径包括NADPH氧化酶的激活和活性氧的生成[29]。这些研究结果表明,血管损伤早期阶段的AF改变是广泛血管壁病变的信号,AF的NADPH氧化酶的激活参与血管重塑的发生发展。
血管炎症:传统观点认为,血管炎症反应是一个“自内而外”的过程,以白细胞/单核细胞为中心,主要发生在内膜。在这一假说中,损伤内膜表达表面黏附分子和炎性介质,吸引单核细胞迁移进入内膜和(或)中膜。然而,新的“从外向内”假说被越来越多的证据所支持,这一假说认为,血管炎症自外膜产生,最终导致中膜和内膜重塑,血管炎症始于外膜[30]。血管外膜不仅是血管炎症的“发源地”,而且是血管炎症发展的“主战场”:外膜含有巨噬细胞和树突细胞等固有免疫细胞和成纤维细胞,后者在一定条件下也可以发挥免疫作用,与固有免疫细胞联合,产生细胞因子、炎症趋化因子和活性氧,参与抗原-抗体反应和T细胞免疫反应。
结语:血管外膜结构复杂、成分多样,分布有成纤维细胞、结缔组织、滋养血管、神经末梢等多种细胞和组织,其不仅具有支撑和营养作用,而且集神经-内分泌-免疫功能于一体,与动脉粥样硬化、高血压以及血管重塑等多种血管损伤的病理过程密切相关。AF是外膜最重要的细胞成分,AF的NADPH氧化酶激活与活性氧生成,参与多种血管损伤的发生发展,是血管损伤的“导火索”。虽然外膜在血管损伤的发生发展过程中具有重要的地位,但针对外膜及AF的药物研究相对匮乏。以NADPH氧化酶、活性氧入手,研究药物对AF和血管外膜的作用及其机制,或可成为早期干预血管损伤的突破口。
[1] Belknap JK, Orton EC, Ensley B, et al. Hypoxia increases bromodeoxyuridine labeling indices in bovine neonatal pulmonary arteries. Am J Respir Cell Mol Biol. 1997, 16: 366-371.
[2] Majesky MW, Dong XR, Hoglund V. The adventitia: a progenitor cell niche for the vessel wall. Cells Tissues Organs, 2012, 195: 73-81.
[3] Ushio-Fukai M, Tang Y, Fukai T, et al. Novel role of gp91phoxcontaining NAD(P)H oxidase in vascular endothelial growth factorinduced signaling and angiogenesis. Circ Res, 2002, 91: 1160-1167.
[4] Wang HD, Xu S, Johns DG, et al. Role of NADPH oxidase in the vascular hypertrophic and oxidative stressresponse to angiotensin II in mice. Circ Res, 2001, 88: 947-953.
[5] Liu J, Ormsby A, Oja-Tebbe N, et al. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res, 2004, 95: 587-594.
[6] Scott NA, Cipolla GD, Ross CE, et al. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation, 1996, 93: 2178-2187.
[7] Wilcox JN, Okamoto EI, Nakahara KI, et al. Perivascular responses after angioplasty which may contribute to postangioplasty restenosis: a role for circulating myofibroblast precursors? Ann N YAcad Sci, 2001, 947: 68-90.
[8] Li G, Chen SJ, Oparil S, et al. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation, 2000, 101: 1362-1365.
[9] 陶婷, 朱鼎良, 龚兰生. 自发性高血压大鼠胸主动脉外膜胶原分布及含量的变化. 高血压杂志, 2001, 9: 50-52.
[10] Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol, 2003, 200: 500-503.
[11] Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen, 2005, 13: 7-12.
[12] Anwar A, Li M, Frid MG, et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol, 2012, 303: L1-L11.
[13] Duque JC, Vazquez-Padron RI. Myofibroblasts: the ideal target to prevent arteriovenous fistula failure? Kidney Int, 2014, 85: 234-236.
[14] Kleniewska P, Piechota A, Skibska B, et al. The NADPH oxidase family and its inhibitors. Arch Immunol Ther Exp (Warsz), 2012, 60: 277-294.
[15] 叶国红, 韩莲花, 李红霞, 等. 探讨血管紧张素II对人脐静脉内皮细胞产生超氧阴离子和1型纤溶酶原激活物抑制剂的作用机制.中国循环杂志, 2012, 27: 141-144.
[16] Altenhöfer S, Kleikers PW, Radermacher KA, et al. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci, 2012, 69: 2327-2343.
[17] Cifuentes ME, Rey FE, Carretero OA, et al. Upregulation of p67phox and gp91phox in aortas from angiotensin II-infused mice. Am J Physiol Heart Circ Physiol, 2000, 279: 2234-2240.
[18] 刘亚洋, 李鹤, 吴宗贵, 等. 晚期糖基化终产物对大鼠血管外膜成纤维细胞中烟酰胺腺嘌呤二核苷酸磷酸氧化酶p22phox亚基及活性氧表达的影响, 中国循环杂志, 2012, 27: 228-231.
[19] Chen F, Barman S, Yu Y. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med, 2014, 73: 201-213.
[20] Li Y, Yan CH, Han YL. CREG mediated adventitial fibroblast
phenotype modulation: a possible therapeutic target for proliferative vascular disease. Med Hypotheses, 2012, 79: 95-97.
[21] Silver FH, Horvath I, Foran DJ. Viscoelasticity of the vessel wall: The role of collagen and elastic fibers. Crit Rev Biomed Eng, 2001, 29: 279-301.
[22] Di Wang H, Hope S, Du Y, et al. Paracrine role of adventitial superoxide anion inmediating spontaneous tone of the isolated rataorta in angiotensin II- induced hypertension. Hypertension, 1999, 33: 1225-1232.
[23] Barman SA, Chen F, Su Y, et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol, 2014, 34: 1704-1715.
[24] Ogeng'o J, Ongeti K, Obimbo M, et al. Features of Atherosclerosis in the Tunica Adventitia of Coronary and Carotid Arteries in a Black Kenyan Population. Anat Res Int, 2014, 2014: 456741.
[25] Siow RC, Mallawaarachchi CM, Weissberg PL. Migration of adventitial myofibroblasts following vascular balloon injury: insights from invivo gene transfer to rat carotid arteries. Cardiovasc Res, 2003, 59: 212-221.
[26] Usui M, Egashira K. Angiotensin II receptor and oxidative stress. Nihon Rinsho. 2002, 60: 1893-1897.
[27] Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc Res, 2007, 75: 679-689.
[28] Arribas SM, Gonzalez C, Graham D, et al. Cellular changes induced by chronic nitric oxide inhibition in intact rat basilar arteries revealed by confocal microscopy. J Hypertens, 1997, 15: 1685-1693.
[29] Shen WL, Gao PJ, Che ZQ. NAD(P)H oxidase-derived reactive oxygen species regulate angiotensin-II induced adventitial fibroblast phenotypic differentiation. Biochem Biophys Res Commun, 2006, 339: 337-343.
[30] Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res,2007,75:640-648.
2014-09-24)
(编辑:王宝茹)
国家自然科学基金资助项目(81273976);中国中医科学院基本科研业务费自主选题项目(ZZ2013002)
100053 北京市,中国中医科学院广安门医院 综合科
王洋 主治医师 博士 主要研究方向为心血管病的中西医结合治疗 Email:wangyangzb11@163.com 通讯作者:雷燕
Email: leiy999@163.com
R54
A
1000-3614(2015)04-0404-04
10.3969/j.issn.1000-3614.2015.04.026
