基于四氮唑配体构筑的配合物研究进展
2015-12-13蔡佳丽董若婷
蔡佳丽,邓 洪 ,董若婷
(华南师范大学化学与环境学院,广州510006)
自从Werner 于1893年创立配位化学以来,对配合物的研究已成为无机化学中最活跃的领域. 化学家利用配位键及各种超分子作用力(例如:氢键、π-π 堆积、范德华作用、疏水亲脂作用、静电作用等),构筑奇特新颖、种类繁多的配合聚合物,并探索它们在化学与材料科学中的潜在应用[1]. 微孔配位聚合物是指无机金属中心或金属簇与有机官能团通过共价键或离子-共价键相互连接,共同构筑具有周期性网络结构且具有规则孔道或者孔穴结构的晶态多孔材料.在网络结构上,与矿物、沸石分子筛和其他无机类沸石分子筛结构相似. 由于它们复合了高分子聚合物和配位化合物两者的特点,在分子与离子交换、吸附与选择性催化、光电子与磁性材料等方面具有潜在的应用价值[2]. 随着以电子、信息、新能源、生物以及新材料等为代表的高新技术日益发展,具有特殊物理、化学和生物化学功能的配合物在国际上更是得到蓬勃的发展.近年来,配位聚合物的晶体工程发展十分迅速,已成为化学的主要研究领域之一.
四氮唑微孔配位聚合物材料具有其特殊的结构,与传统的微孔材料活性炭、沸石材料、硅酸盐、硅铝酸盐、磷铝酸盐等相比,四氮唑微孔配位聚合物具有独特的优势:一方面,可以将金属离子所特有的光、电、磁学等特性引入合成的配位聚合物中;另一方面,有机配体具有多样性、可修饰性,可与各种金属离子形成不同组合,为设计合成尺寸可控、形状可控和性质可控的各种配位聚合物提供了可能. 由于四氮唑微孔配位聚合物的孔结构高度有序,孔尺寸可控和孔表面的官能团和表面势能可控制,因此它们在分子识别、气体储存、多相催化、非线性光学等方面的具有潜在应用价值[3]. 近年来,设计合成四氮唑微孔配位聚合物,开发该类配合物的功能复合化、高性能化在材料科学领域成为了热点[4].
1 四氮唑配体的配位模式
四氮唑配体具有多配位点,是一种多齿配体,具有多种配位模式.它既可以作为螯合配体,也可以作为桥联配体,为配合物的世界增添了许多新颖结构,有“万能配体”的美誉[5].自从引入5-取代基于四氮唑配合物上,四氮唑配合物的结构有了极大的丰富,并且构筑了更多灵活多样、形式各异的配位模式,如图1 所示.

图1 四氮唑环的配位模式Figure 1 Coordination modes of tetrazole ligand
2 四氮唑配位物的合成
四氮唑配合物合成方法主要包括原位合成和使用配体合成2 种方法.最近,作者采用过渡金属盐为催化剂,在水热反应条件下,利用叠氮化物和不同取代基的氰化物在水相中反应,原位一步合成了系列5-取代基四氮唑配合物[6-14].这种[2 +3]环加成方法方法简便、原料易得、成本降低的优点,该方法报道以后立即引起了同行的广泛关注,并使得不同有机腈原位合成5-取代四氮唑的理论和研究有了进一步的发展.
2.1 具有新颖拓扑结构四氮唑配合物的合成
本课题组合成了具有高对称性的具有金属有机配合物[Cd15(5MT)18(OH)4(SO4)4](5MT=5-methyl-1H-tetrazole).该配合物由罕见的Cd3(μ3-OH)(μ3-SO4)核和2 种不同配位模式的四氮唑配体共同构成,呈现了一个非常新颖的39·412·515拓扑结构[6].尤为新颖的是,它在脱水和吸水过程中会产生体积收缩和膨胀的“呼吸”效应,并在收缩和膨胀过程中化合物的荧光也发生了变化(图2)[6].
2.2 引入辅助配体和阴离子调控配合物结构
辅助配体和阴离子小分子对配合物的分子结构构筑也起了非常关键的作用.课题组以喹啉基取代的四氮唑为主配体,以多酸类的配体为辅助配体,合成了6个结构新颖的金属有机配合物.[Cd(QCT)2](1),[Cd(QCT)N3·H2O](2),[Zn(QCT)Cl](3),[Cd2(QCT)2(IN)2]·H2O (4),[Cd2(QCT)(3BC)·2(H2O)]·2H2O (5),2[Cd3(QCT)2(4BC)·2(H2O)]·6H2O (6)(QCT =5-喹啉基取代四氮唑,IN = 异烟酸,3BC=均苯三甲酸,4BC =均苯四甲酸)(图3).进一步证明了不同小分子的引入,如:叠氮根、卤素离子、异烟酸、均苯三甲酸以及均苯四甲酸对金属有机配位框架的形成和调控影响非常大[7].

图2 配合物在脱水和吸水过程中会产生体积收缩和膨胀Figure 2 Volume contraction or expansion of the complexes in the process of dehydration or water absorption

图3 配合物1~6 合成路线图Figure 3 Preparation details of Compounds 1-6
另外,通过选择不同的取代基烷基有机腈,改变不同反应的阴离子,原位合成了系列具有新颖结构的不同5-取代基的同核金属四氮唑配位聚合物(图4)并研究了它们的特异荧光性质[8].
2.3 利用双腈配体原位合成双四氮唑配合物
作为之前四氮唑配合物研究工作的延续,课题组选择采用了丙二腈、丁二腈作为合成四氮唑的前驱物,在水热条件下与叠氮化钠、金属Zn(Ⅱ)/Cd(Ⅱ)/Mn(Ⅱ)盐反应,原位合成金属有机框架. 对于此前驱物配体的选择主要是基于以下几点考虑:二腈有2个氰基,在该实验条件下,它的产物可以存在3 种可能:(1)2个氰基直接水解成2个羧基生成的是HOOC—(CH2)n—COOH(n =1);(2)它可以只有一端的氰基发生[2 +3]环加成反应,而另一端而水解形成—COOH,则相应的产物为1H-四氮唑-5-乙酸(1H-tetrazolate-5-carboxylate),它具有6个潜在的配位点(4个N 原子和2个O 原子),而目前对于该种配体的报道尚少,尤其是基于1H-四氮唑-5-乙酸的金属配合物的研究. (3)2个氰基都发生[2 +3]环加成反应生成双四氮唑,对应的产物为双(四氮唑基)甲烷(bis(tetrazole)methane),而关于双四氮唑的研究报道还非常有限[9];配体中存在的—CH2—可以增加结构的柔性.

图4 配合物的合成路线及其配位模式[8]Figure 4 In situ hydrothermal syntheses of the compounds and their coordination modes[8]
我们证明了对于具有2个氰基的前驱物,在原位合成四氮唑时,可以通过调节加入的叠氮的量来控制合成只含有1个四氮唑和双四氮唑的金属配合物;另外通过调节pH 值,调控形成双四氮唑或配体一端是四氮唑另一端是羧基的双功能配合物(图5)[10].
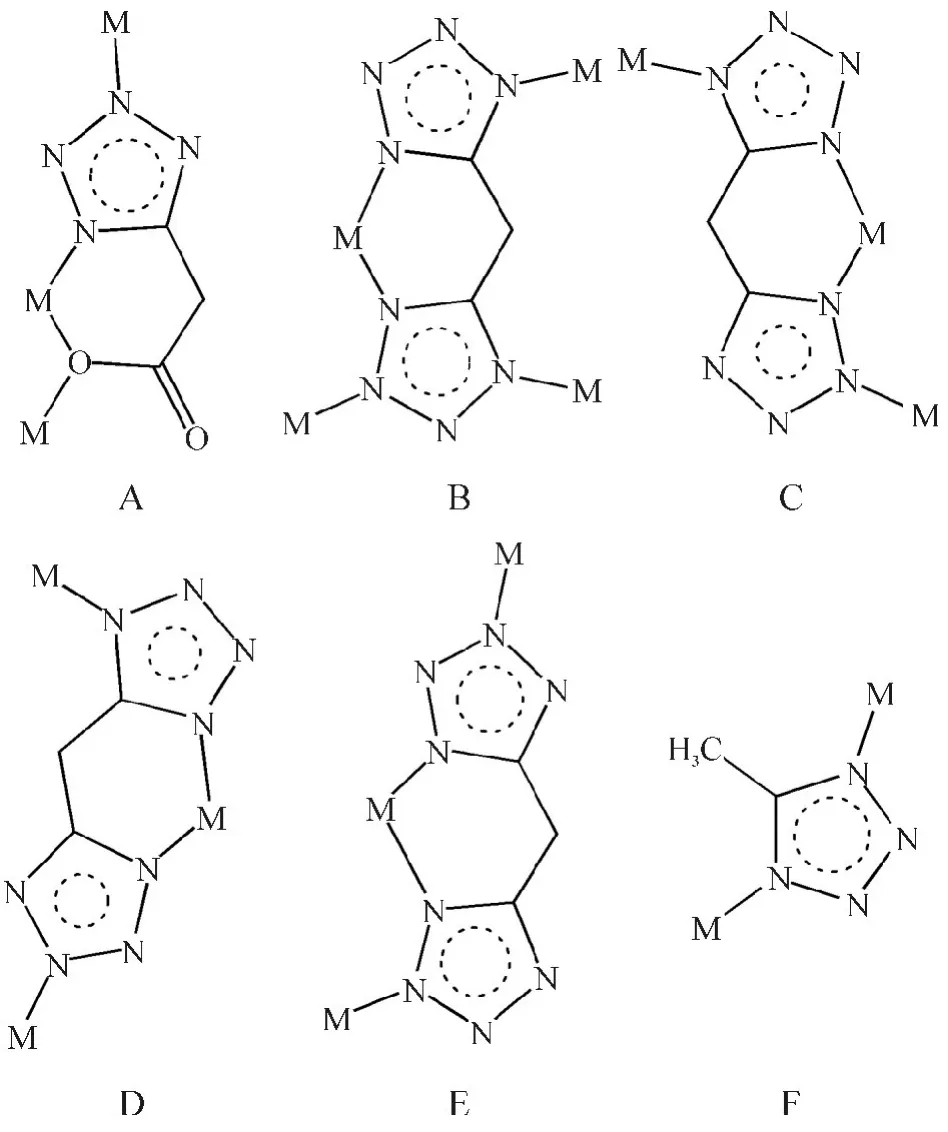
图5 配合物的配体所呈现的配位模式图Figure 5 Coordination models of the complexes
2.4 原位合成异核四氮唑配合物
2.4.1 合成过渡金属异核四氮唑配合物 已有大量的四氮唑化合物采用水热或溶剂热反应原位合成,主要集中在单核四氮唑配合物的制备上,而对异核或多核四氮唑配合物的合成规律的探索很少涉猎.事实上,异核或者多核四氮唑类配合物在吸附、电导、光学材料、磁性能方面具有潜在的应用价值,而且在异核或多核体系中,不同的金属阳离子在与四氮唑上的氮原子配位的优先权及差异性为配合物的结构多样性提供更广阔的空间.
课题组在四氮唑配合物的水热或溶剂热反应中选择CuI 作为路易斯酸盐,同时加入含碱金属的小分子盐(KCl/NaCl).其中选择CuI,主要是基于Cu+阳离子与I-阴离子超强的结合能力可以用来构筑次级结构单元(secondary building units,SBU). 在以乙腈或者苯作为溶剂的条件下,乙腈、NaN3、CuI 和不同的碱金属盐通过原位反应生成不同的Copper(I)-Alkaline 异核四氮唑配合物(图6)[11]. 单晶衍射显示四氮唑环上的氮原子去质子化,而且所有的氮原子都和金属配位. 进一步说明了四氮唑环作为多齿配体的超强配位能力. 配合物[Cu2K(Mtta)I2]是由少见的5 桥联的Mtta 配体与Cu-I-K 层构筑而成;配合物[Cu3K(Mtta)3I]是无数个无机阳离子簇[Cu6I2]4+与4 桥联的Mtta 配体构筑而成;配合物[Cu2Na(Mtta)2(CH3CN)I]是由开放的子网络Cu(Mtta)与Na+构筑而成.结果对Demko-Sharpless 关于四氮唑合成理论在异核领域作了近一步的补充,作为催化剂的路易斯酸盐中金属在与碱金属盐共存时,具有配位优先性,而且尽可能地占据最低能量位阻位置,即采用1-配位模式. 此方法为合成其他具有特殊功能的高维异核四氮唑化合物提供了一个新思路.

图6 原位溶剂热合成异核配合物[11]Figure 6 In situ solvothermal syntheses of the complexes[11]
2.4.2 合成稀土-过渡金属异核四氮唑配合物目前报道的四氮唑配合物结构中,中心金属通过原位合成的3d-4f 异核金属配合物尚无报道. 众所周知,3d-4f 异核金属配合物不仅拥有迷人的结构和拓扑,而且在催化、气体吸附、磁性和光学上都有潜在的应用.然而,由于稀土离子变化的配位模式和高的配位数,同时在过渡和稀土离子之间存在竞争反应,因此对于3d-4f 异核金属配合物的合成仍然是一项挑战.选择适当的配体是非常重要的. 因此课题组选择3-氰基苯甲酸作为前躯体来构筑3d-4f 异核四氮唑配合物,原因如下:(1)3-氰基苯甲酸通过原位水热/溶剂热合成四氮唑苯甲酸配体(3-H2tzba);(2)3-H2tzba 是一个潜在的多齿配体,它同时含有氮原子和氧原子,根据软硬酸碱理论,四氮唑上的氮原子倾向于和过渡金属离子配位,羧基上的氧原子倾向于和稀土离子配位.
以3-氰基苯甲酸作为前躯体合成了一系列3d-4f 异核金属配合物,即[LnCu(3-tzba)2(H2O)4][Ln = Eu(7);Gd(8);Tb(9);Dy(10);Ho(11);Nd(12);Sm(13);Pr(14);3-tzba =5-苯甲酸基取代四氮唑(图7).并用X 射线单晶衍射、红外光谱、元素分析、部分物质用荧光光谱、磁性分析等手段对这些化合物进行了表征. 这是课题组首次报道的通过原位合成得到异核四氮唑配合物[12].由于稀土元素在光、电、磁等方面独特的性质,在信息、生物、新材料、新能源、空间和海洋等科技群中有广阔的应用前景,被誉为新材料的宝库. 稀土和过渡金属通过有机配体桥连得到的杂核配合物具有稀土和过渡金属的双重性质,因此由于其含有不同的配位中心稀土-过渡金属异核配合物可形成具有优于同核配合物的化学物理性能.

图7 配合物[LnCu(3-tzba)2(H2O)4]的合成示意图Figure 7 In situ hydrothermal synthesis of[LnCu(3-tzba)2(H2O)4]
3 四氮唑配合物的应用研究
3.1 荧光探针
四氮唑化合物都有芳香环,与过渡金属或稀土金属配位的四氮唑配位聚合物大部分有较强的荧光.其中,四氮唑配合物电子吸收的主要类型有以下几种:配体内部跃迁、金属中心跃迁、配体到金属电荷跃迁和金属到配体电荷跃迁.
本课题组选用乙腈和金属Cd(II)盐原位合成四氮唑配合物[Cd(μ2-Cl)(μ4-5MT)]n(5MT =5-methyl-1H-tetrazole).在水溶液或DMF 中,该化合物能对进行选择性的吸附,并且随着浓度的增加,其荧光强度显著下降,因此该四氮唑多孔配位聚合物可用于致癌物质NaNO2的检测.显示出该配合物在荧光探针方面具有潜在的应用价值(图8)[13].
考虑Zn 配合物具有良好的发光性能,课题组利用5-脂肪链取代四氮唑类配体和异烟酸构筑了不同构型的Zn 金属有机配位框架,并针对其荧光性能在室温下对该系列配合物进行了研究. 当配合物分别在356、331、370 和357 nm 光的激发下,在397、443、447 和410 nm 处出现宽峰.配合物的荧光寿命分别为:12.35、10.43、3. 58 和4.86 ns. 根据文献[14]报道,室温下四氮唑配体在325 nm 处发出强的荧光.因此,配合物发出的强荧光可以归因于配体到金属的电荷转移(LMCT),而不同5 位取代基导致了荧光性能的变化[14].
3.2 催化及分子磁体
单分子磁体是指那些在磁场下能够被磁化,当磁场去除后仍能保持磁性的单分子,其磁化强度对外磁场的曲线会出现磁滞回线[15].分子磁体由于具有结构多样、密度小、可塑性和透光性好、易于加工成型等许多更优越的性能而受到人们的重视. 到目前为止,已经有不少稀土单分子磁体的报道[16-18],但未曾有稀土四氮唑配合物分子基磁体报道.
近年来,在超分子化学和晶体工程领域,通过水热合成四氮唑配合物受到越来越多的关注.一般,四氮唑配合物是通过腈和叠氮[2 +3]环加成得到的,以路易斯酸盐为催化剂.到目前为止,大部分文献报道的配合物都是以路易斯酸盐为催化剂,例如Ag(Ⅰ)、Cu(Ⅰ)/(Ⅱ)、Zn(Ⅱ)和Cd(Ⅱ)等. 我们通过稀土离子Ln(Ⅲ)代替传统的过渡金属离子作为催化剂,利用3-氰基苯甲酸与叠氮钠原位水热合成了一系列四氮唑配合物.分子式通式为[Ln2(3-Htzba)2(3-tzba)2(H2O)8]·4H2O [Ln = Gd,1;Dy,2;Eu,3;Tb,4;Sm,5;Er,6;3-H2tzba =3-(1Htetrazol-5-yl)benzoic acid],并用红外光谱、元素分析对这些化合物进行表征,X 射线单晶衍射分析表明这些配位聚合物通过配体上的羧基连接2个稀土离子形成了一系列二核的零维结构[19].单分子磁体行为的一个重要判据就是交流磁化率出现频率变化的虚部信号(χ″),即慢磁弛豫现象. 故我们对配合物2 的交流磁化率进行了探讨,研究了不同频率下交流磁化率随温度的变化及不同温度下交流磁化率随频率的变化(图9).这些数据说明了磁化强度的慢弛豫现象的存在,具有明显单分子磁体特征.在零静磁场时,χ″-T 图中,虚部磁化率在不同频率下都出现了极大值,低于4 K 时,配合物2 的实部和虚部磁化率具有很强的频率依赖性,随着温度升高,磁化率也增大,高于4 K 时,随着温度升高,磁化率降低.同样,在χ″-ν 图中,虚部磁化率在不同温度下也出现了极大值,在温度高于3 K 时,磁化率峰值出现在约20 Hz.

图8 (A)配合物浓度为1×10-4 mol /L DMF 中比较不同阴离子的荧光强度;(B)配合物荧光强度随离子浓度的增加而降低Figure 8 (A)Comparison of the luminescence intensity of the complex in 1 ×10 -4 mol/L DMF solutions with different ions of selected salts,(B)Excitation and photoluminescence spectra of the complex in the solid state and PL spectra of complex in DMF solutions with different concentrations of NaNO2(excited and monitored at 236 nm and 370 nm,respectively)

图9 不同频率下实部和虚部交流磁化率随温度的变化(A、B)及不同温度下实部和虚部交流磁化率随频率的变化(C、D)Figure 9 Temperature (A,B)and frequency (C,D)dependence of the in-phase (χ',left)and the out-of-phase (χ″,right)ac magnetic susceptibility under a zero-dc field for complex
进一步探讨动态磁场,在1.8 K 和3.8 K 下,为了减小量子弛豫,外加直流场测出其磁化率对频率的曲线(图10).在1.8 K 下,随着外加场强的增加,磁化率的峰值迅速向低频率区域转换,然而在3.8 K下,外加场强3 000 Oe,磁化率的峰值慢慢的向低频率区域转换,说明在1.8 K 直流场下,弛豫时间明显的增强了,在3.8 K 下有微小的增强.
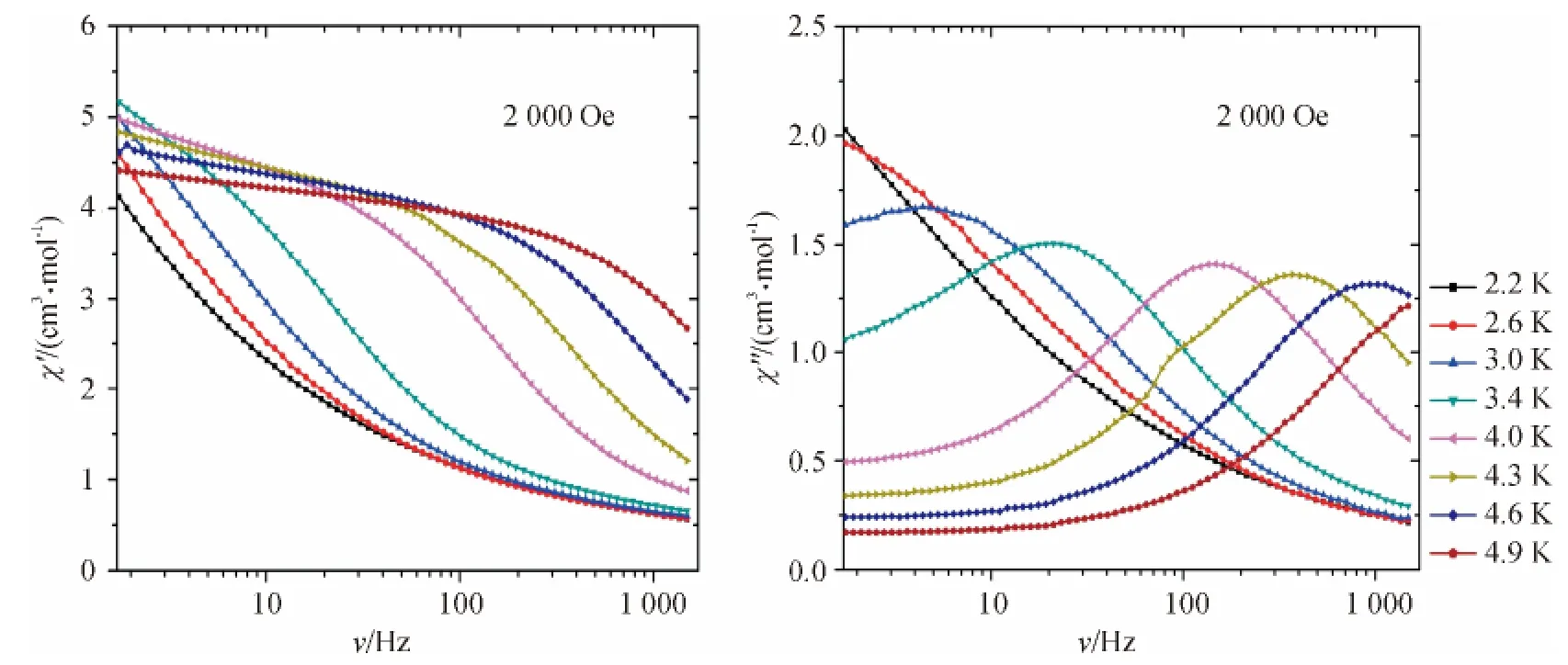
图10 2 000 Oe 场强下实部和虚部交流磁化率随频率的变化Figure 10 Frequency dependence at 2 000 Oe of the in-phase (χ',left)and the out-of-phase (χ″,right)ac magnetic susceptibility at different temperatures
进而,选择场强为2 000 Oe 在不同温度下,磁化率对温度的曲线(图11). 温度高于4.0 K 时,符合Arrhenius 定律,得出弛豫时间τ =1.1 ×10-9s,各向异性有效能垒Δeff =54.8 K,这与在零场下得出的结果基本是一致的. 然而量子弛豫时间大约是0.1 s,这比零场下得出的结果大. 这是第一例报道的通过原位水热合成的单分子磁体.

图11 不同温度下的Cole-Cole 图Figure 11 Cole-Cole plots using the ac susceptibility data
3.3 吸附性质
最近,本课题组合成了新型四氮唑多孔配位聚合物:[Cd(μ2-Cl)(μ4-5MT)]n(5MT =5-甲基-1H-四氮唑).该配合物同时表现了较好的气体吸附性能,在77 K 和1个大气压条件下,对氢气的吸附能力达到了0.67%(质量比,全文同),相当于被广泛应用于石油化工、煤化工的ZSM-5 型沸石的吸附能力(0.7%)[13].
此外,我们还选择四氮唑-1-乙酸作为配体构筑出新颖微孔金属有机配位框架(图13-14),主要基于以下几点考虑.首先四氮唑-1-乙酸既含有四氮唑环又含有羧酸根.四氮唑环具有4个芳香氮原子,所以四氮唑类配体具有多个配位点,拥有多种多样的配位模式,可以用来构筑多核单元或者高维结构.其次,羧酸根离子是以配位模式多样、配位能力强、在配合物自组装中作用重大而闻名的.加之,四氮唑环和羧酸根之间—CH2—官能团的引入,增加了配体的柔性,使得其配位模式更是多样化.
4 展望
要研制理想的微孔配位聚合物荧光探针,首先要满足几点要求:(1)必须对被分析物有明显不同的荧光响应;(2)荧光响应的不同必须是对特定的被分析物;(3)响应最好是可逆的(但不对所有情况适用);(4)荧光响应的不同必须在可检测范围内.
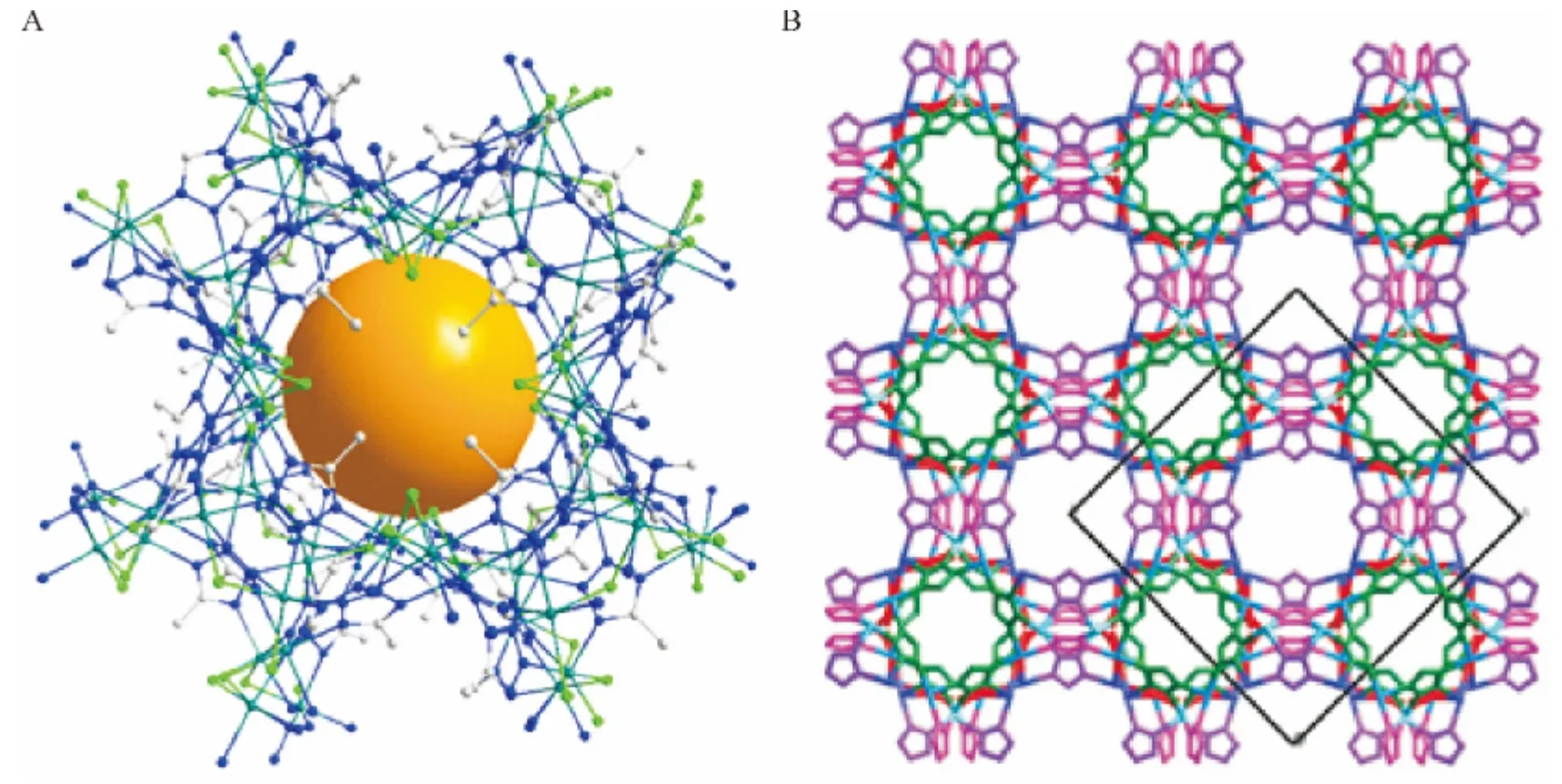
图12 (A)局部显示配合物的微孔结构、(B)沿C 轴显示配合物的三维微孔结构Figure 12 (A)part of its microporous framework showing one of the large voids. Each crystallographically-unique Cd and 5MT moiety is coloured differently and (B)the 3D microporous framework viewed along the c-axis

图13 配合物铜离子配位环境图Figure 13 Coordination environments of copper atoms in complex
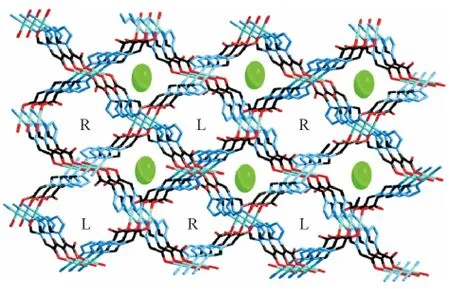
图14 配合物的三维结构图Figure 14 Perspective view of the porous 3D network in the complex with perchlorate ions (spheres)in one channel and helical water chains in the other channel (L & R)
相比其他荧光探针材料,微孔配位聚合物具有明显的优势:首先,微孔配位聚合物兼备有机物与无机物特性,合成方法多样,且具有较高的比表面积和良好的结构灵活性. 基于微孔配位聚合物材料的这些特征,通过改变生色团类型、次级建筑单元及其相对空间排列,可以改变微孔配位聚合物的电子特性,合成出不同用途的荧光材料;其次,微孔配位聚合物由于有开放的配位点,能够可逆地键合目标分析物,发光中心的协同定位可以提供一个传感器的转导机制.最后,由于微孔配位聚合物的结构灵活性可以导致物质配位环境及发光性能发生戏剧性的变化,因此可用于分子或离子检测.
目前对微孔配位聚合物材料作为荧光探针性能的研究处于初级发展阶段,绝大多数的文献仅展示了配合物的荧光光谱,对客体分子导致荧光响应的讨论和合理设计微孔配位聚合物作为荧光探针的应用还处于起步和探索阶段,仍存在一些科学问题有待解决.
四氮唑环具有芳香性,是个很好的生色团. 锌、镉等过渡金属四氮唑配合物都有四氮唑环具有芳香性,是很好的生色团. 锌、镉等过渡金属四氮唑配合物都有较强的荧光.四氮唑环上的配位点多,极有可能存在未配位的氮原子,即为路易斯碱,其与氢离子、金属离子等路易斯酸互相作用,易导致配合物的荧光响应发生变化,是pH 探针、离子探针材料的理想选择.基于这些考虑,作者认为锌、镉等过渡金属四氮唑配体类的微孔配位聚合物,很可能是作为微孔配位聚合物荧光探针材料的理想选择. 我们的初步研究成果已经初步证实了这个设计的合理性和可行性[13].
总之,微孔配位聚合物被关注的重点在于组装具有新颖结构和功能的研究上. 物质的结构和组成所决定了它们的性质,设计和合成微孔配位聚合物成为当前具有挑战性的课题. 通过改变不同的四氮唑5 位取代基和金属离子的调控,完成对四氮唑配位聚合物的设计、合成,实现包括微孔结构的孔径控制合成,在对材料尺寸及维度进行限制的基础上,研究在限制空间的条件下(多孔配位聚合物的孔道)和作用力(氢键、静电、疏水作用等),配位聚合物与小分子或阴、阳离子作用之间的能量传递及光电性质的变化规律,同时系统研究其对各种气体,特别是能源气体(如氢气、甲烷)的吸附性能,充分发掘新颖四氮唑微孔配位聚合物的荧光探针和高气体吸附性能材料的潜能,为开发具有复合功能的四氮唑微孔配位聚合物及应用提供理论依据和合适的新材料体系.
[1]洪茂椿,陈荣,梁文平. 21 世纪的无机化学[M]. 北京:科学出版社,2005.
[2]游效曾,分子材料-光电功能化合物[M]. 上海:上海科学技术出版社,2001.
[3]Zhang J P,Zhang Y B,Lin J B,et al. Metal azolate frameworks:From crystal engineering to functional materials[J]. Chemical Reviews,2011,112(2):1001-1033.
[4]Aromí G,Barrios L A,Roubeau O,et al. Triazoles and tetrazoles:Prime ligands to generate remarkable coordination materials[J]. Coordination Chemistry Reviews,2011,255(5):485-546.
[5]Dincǎ M,Long J R. High-enthalpy hydrogen adsorption in cation-exchanged variants of the microporous metal-organic framework Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2[J]. Journal of the American Chemical Society,2007,129(36):11172-11176.
[6]Deng H,Qiu Y C,Li Y H,et al. Reversible shrinkage and expansion of a blue photofluorescent cadmium coordination polymer and in situ tetrazole ligand synthesis[J].Chemical Communications,2008(19):2239-2241.
[7]Ma L,Yu N,Chen S,et al. Construction of diverse Cd II/Zn II coordination polymers based on 5-(quinolyl)tetrazolate generated via in situ hydrothermal synthesis[J].Crystal Engineering Communication,2013,15(7):1352-1364.
[8]Qiu Y,Li Y,Peng G,et al. Cadmium metal-directed three-dimensional coordination polymers:In situ tetrazole ligand synthesis,structures,and luminescent properties[J]. Crystal Growth & Design,2010,10(3):1332-1340.
[9]Li Y W,Chen W L,Wang Y H,et al. Entangled zincditetrazolate frameworks involving in situ ligand synthesis and topological modulation by various secondary N-donor ligands[J]. Journal of Solid State Chemistry,2009,182(4):736-743.
[10]Sun L,Ma L,Cai J B,et al. Novel tetrazole-based metal-organic frameworks constructed from in situ synthesize bifunctional ligands:Syntheses,structure and luminescent properties[J]. Crystal Engineering Communication,2012,14(3):890-898.
[11]Liu B,Qiu Y C,Peng G,et al. In situ solvothermal syntheses of a heteronuclear copper (I)-alkaline metallic tetrazole-based coordination polymer[J]. Crystal Engineering Communication,2010,12(1):270-276.
[12]Liang L,Peng G,Ma L,et al. A new family of 3d-4f heterometallic tetrazole-based coordination frameworks:In situ tetrazole ligand synthesis,structure,luminescence,and magnetic properties[J]. Crystal Growth & Design,2012,12(3):1151-1158.
[13]Qiu Y,Deng H,Mou J,et al. In situ tetrazole ligand synthesis leading to a microporous cadmium-organic framework for selective ion sensing[J]. Chemical Communications,2009 (36):5415-5417.
[14]Ma L,Qiu Y C,Peng G,et al. In situ tetrazole ligand synthesis of two-fold interpenetrating zinc coordination frameworks[J]. European Journal of Inorganic Chemistry,2011,2011(23):3446-3453.
[15]Yan Z,Li M,Gao H L,et al. High-spin versus spincrossover versus low-spin:Geometry intervention in cooperativity in a 3D polymorphic iron (ii)-tetrazole MOFs system[J]. Chemical Communications,2012,48(33):3960-3962.
[16]Woodruff D N,Winpenny R E P,Layfield R A. Lanthanide single-molecule magnets[J]. Chemical Reviews,2013,113(7):5110-5148.
[17]Guo Y N,Xu G F,Wernsdorfer W,et al. Strong axiality and Ising exchange interaction suppress zero-field tunneling of magnetization of an asymmetric Dy2single-molecule magnet[J]. Journal of the American Chemical Society,2011,133(31):11948-11951.
[18]Jiang S D,Wang B W,Su G,et al. A mononuclear dysprosium complex featuring single-molecule-magnet behavior[J]. Angewandte Chemie,2010,122(41):7610-7613.
[19]Liang L,Peng G,Li G,et al. In situ hydrothermal synthesis of dysprosium (Ⅲ)single-molecule magnet with lanthanide salt as catalyst[J]. Dalton Transactions,2012,41(19):5816-5823.
