Pt负载钙钛石催化剂上氧空位对CO氧化的促进作用
2015-11-30孔令智王谦徐丽闫永胜李华明杨向光
孔令智王谦徐丽闫永胜李华明*,杨向光
(1江苏大学能源与动力工程学院,镇江212013)
(2江苏大学能源研究院,镇江212013)
(3中国科学院长春应用化学研究所,长春130022)
Pt负载钙钛石催化剂上氧空位对CO氧化的促进作用
孔令智1王谦1徐丽2闫永胜2李华明*,2杨向光*,3
(1江苏大学能源与动力工程学院,镇江212013)
(2江苏大学能源研究院,镇江212013)
(3中国科学院长春应用化学研究所,长春130022)
采用柠檬酸法制备了LaMnO3、LaFeO3、La0.5Sr0.5MnO3、La0.5Sr0.5FeO3,通过负载纳米Pt合成了Pt负载钙钛石催化剂,XRD与IR数据表明合成的催化剂具有钙钛石相,TEM数据表明合成的纳米Pt粒径为~3 nm,均匀分散在钙钛石上。在CO氧化反应中,发现钙钛石的氧化-还原性能是影响其活性的重要因素,因而,Mn系钙钛石表现出较高的CO氧化活性。负载纳米Pt后,Fe系钙钛石则显示出更优异的CO氧化活性,CO完全转化的温度从350℃降至120℃。吸附实验表明钙钛石上氧空位对促进O2的吸附起着非常重要的作用,也是影响CO低温氧化的重要因素之一。
钙钛石;纳米Pt;氧化-还原;氧吸附
CO氧化在燃料电池、CO气体传感器、CO2激光器、空气净化及封闭系统内微量CO消除等方面有着非常广泛的应用[1]。特别是在汽车尾气净化中,CO氧化过程同时还影响着NO还原的过程。CO氧化反应还因其过程没有选择性问题常被用作模型反应来研究催化剂的催化行为。从已采用的催化体系看,可分为非贵金属催化剂和贵金属催化剂[2-4]。对于贵金属催化体系,用于CO催化氧化的催化剂主要是负载型贵金属(Pt、Pd、Au)催化剂。最具代表性的体系是Au/MO,可以在低于室温条件下催化氧化CO,其性能不仅与Au的粒径有关[5],也与载体种类有关[3]。在负载型催化体系中,CeO2由于具有储放氧功能,经常被选择作为载体。Fernández-García等[6]发现在Pd/CexZr1-xO2/Al2O3上CO催化氧化活性明显高于Pd/Al2O3催化剂,将其归因于引入Ce后,在Pd-Ce界面上生成了空穴,促进了CO和O2在Pd-Ce界面上的活化,从而提高了催化剂的活性。
对非贵金属体系,用于CO催化氧化的催化剂主要是过渡金属氧化物(CuO、Co3O4)催化剂等。在Co3O4体系中,申文杰等[7]采用溶剂热法实现了氧化物的形貌的调控,进而发现110晶面显示出非常高的活性,结合理论模型指出该晶面可以大量的暴露Co3+离子,而Co3+离子是CO氧化的活性中心。王成等[8]也发现在纳米Cu2O体系,不同晶面对CO氧化活性差别较大,高米勒指数晶面活性较高。在Cu-Ce体系的研究中,董林等[9]认为Cu与Ce之间形成的氧空位易于氧的活化,进而增加了CO氧化的活性。虽然对CO氧化反应已进行了非常广泛的研究,并取得显著的进展,但因反应体系复杂,一些影响CO氧化深层次因素还不十分清楚。最近,基于谱学技术的快速发展和理论计算方法的进步,对CO在催化剂表面反应历程有了更深入的了解的。2011年,Yates等[10]在对Au/TiO2进行了详细的研究后,指出随着温度的升高CO在TiO2上的吸附强度明显减弱,因而CO容易在TiO2表面进行氧化反应,而氧的活化易发生在Au与TiO2界面,这一结论得到了理论计算结果的验证。2010年,包信和等[11]发现在金属Pt表面引入FeO1-x,可以显著增加CO的氧化活性,通过理论计算指出活性增加原因主要来自FeO1-x的引入,其作用是提高了催化剂对氧的吸附能力,同时减弱了对CO的吸附能力。上述理论研究表明,对CO催化氧化反应而言,除了催化剂的氧化-还原性能外,氧空位对O2吸附与活化是至关重要的。钙钛石型复合物既具有非常优异的完全氧化活性又具有非常高的热稳定性,还因其结构明确经常被选作模型催化剂,钙钛石型复合氧化物的特殊结构在于通过A位掺杂可调变B位离子的价态和氧空位的含量[12]。
本文选择了氧空位可调的Fe系钙钛石和缺少氧空位的Mn系钙钛石,与纳米Pt复合构成了纳米Pt-钙钛石型催化剂,进而考察了氧空位对CO氧化过程的影响。
1 实验部分
1.1 催化剂的制备
LaMnO3的制备:采用柠檬酸爆炸法合成。一定量的La(NO3)3·6H2O、Mn(NO3)2溶于柠檬酸溶液中。为了保证配位完全,柠檬酸过量10wt%。将上述溶液搅拌蒸干得前驱体。再在300℃下分解,然后700℃煅烧5 h。
LaFeO3及Sr掺杂的钙钛石催化剂的合成采用与LaMnO3相同的制备方法。
Pt胶体的制备:将一定量的表面活性剂PVP (K30)溶于氯铂酸的乙二醇水混合溶液中,加热回流1.5 h,冷却,加入丙酮,离心得到黑色的沉淀,加乙醇超声分散得到Pt胶体。
1wt%Pt/负载钙钛石催化剂制备:取所需的Pt胶体,加入一定量的钙钛石,超声0.5 h,过量的乙醇采用旋转蒸发仪蒸干,80℃干燥过夜,然后在马弗炉中450℃煅烧2 h。
1.2 催化剂的表征
样品粉末X射线衍射(PXRD)实验测试条件:Cu Kα(λ=0.154 06 nm),扫描范围10°~80°,扫描速率10℃·min-1;红外光谱是在德国BRUKER Vertex 70 FTIR光谱仪上进行的,采用KBr压片;氢气程序升温还原(H2-TPR)实验是在自制的仪器上完成的,样品用量为50 mg,在空气气氛下500℃处理0.5 h后降至室温,之后在5%H2/Ar气氛中程序升温还原,用TCD检测器进行记录;TEM表征在日本电子JEOL JEM-2010透射电子显微镜上进行,操作电压为200 kV;样品的BET比表面积和孔径分布在ASAP2010型物理吸附仪(Micromeritics公司)上采用N2吸附法测定,吸附前样品经250℃抽真空(<61.67 Pa)处理3 h。
催化剂表面上CO,O2的吸附及反应性能在自制装置上进行,以高纯Ar为载气,流速30 mL·min-1,催化剂用量50 mg。样品在400℃处理0.5 h后,冷却至室温,用10%O2-Ar吸附0.5 h,经Ar气吹扫以除去物理吸附的O2后,再通入1%CO,同时用QIC-20质谱(Hiden公司)进行检测。
1.3 催化剂的活性评价
CO氧化活性测试在常压条件下固定床反应器中进行的,催化剂50 mg(40~60 mesh),用石英砂(40~60 mesh)稀释,反应气组成为1%CO-99%空气,空速30 000 mL·g-1·h-1,反应尾气用岛津GC-8A型气相色谱仪在线分析检测。
2 结果与讨论
2.1 XRD结构分析
图1为钙钛石和1%Pt负载钙钛石的XRD衍射图。由图中可见,所有的复合氧化物均显示出ABO3的结构特征,合成的LaMnO3和Pt/LaMnO3钙钛石催化剂的特征峰和LaMnO3标准图(JCPDS No. 75-0440)的特征峰一致,LaFeO3和Pt/LaFeO3钙钛石催化剂的特征峰和LaFeO3标准图(JCPDS No.37-1493)的特征峰一致,而Sr掺杂的钙钛石样品则出现了少量SrCO3(JCPDS No.05-0418)的衍射峰,如图1所示。所有Pt负载的钙钛石催化剂均未出现Pt的特征衍射峰,一方面说明金属Pt的含量低,一方面说明负载在钙钛石上的金属Pt纳米粒子是高度分散的,结晶度也不高。
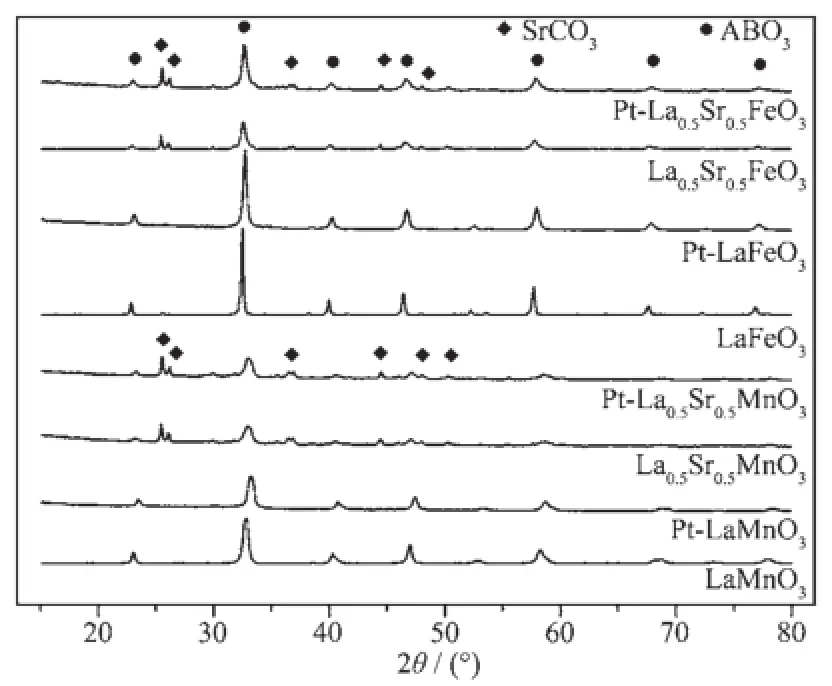
图1 不同钙钛石及其负载Pt催化剂的XRD图Fig.1 XRD patterns for various ABO3mixed oxdes and Pt/ABO3
2.2 IR分析
钙钛石晶体结构是BO6八面体通过共顶点连接而成的,A位阳离子处于BO6八面体之间的间隙中。BO6八面体分子有六个简正振动模式,只有ν3和ν4振动是有红外活性的,B-O键伸缩振动峰(ν3)出现在600 cm-1附近,B-O键弯曲振动峰(ν4)出现在在400 cm-1附近[13]。图2为不同钙钛石及其负载Pt样品的红外吸收光谱。由图可见,对所有样品在600 cm-1附近均出现了钙钛石结构的特征峰。负载Pt后,红外谱图没有发生变化,说明负载过程对钙钛石的结构没有产生明显的影响。然而随着Sr掺杂后,858和1 460 cm-1附近的出现了吸收峰,该吸收峰可归属为SrCO3中碳酸根的振动峰[14]。
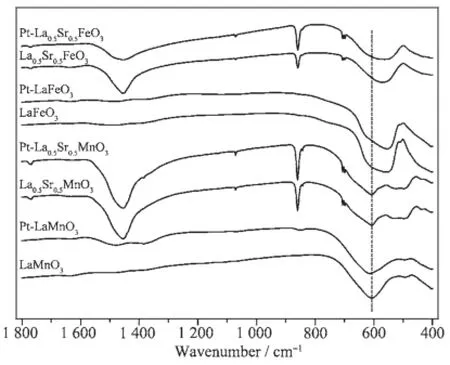
图2 不同钙钛石及其负载Pt催化剂的IR谱Fig.2 IR spectra for various ABO3mixed oxdes and Pt/ABO3
2.3 TEM分析
图3为制备的Pt纳米粒子以及Pt负载钙钛石样品的TEM图。从图3e中可知,合成的Pt纳米粒子比较均匀,平均粒径为~3 nm,并且颗粒呈分散性,无明显的团聚现象。Pt纳米粒子经负载过程后,均匀地分散在钙钛石载体上,粒径仍为~3 nm,煅烧后Pt纳米粒子没有团聚长大,但Pt纳米粒子分散程度随载体的种类不同会产生较大的差异。在LaMnO3上,观察到的Pt纳米粒子较少,可能存在部分团聚的Pt纳米粒子。在La0.5Sr0.5MnO3上,观察到的Pt粒子有所增加,但与Fe系钙钛石相比,观察到的Pt粒子要少些。虽然在Fe系钙钛石上,观察到的Pt纳米粒子数量较多,但分散均匀。对Mn体系而言,许多研究[12,15]均证实Mn系钙钛石几乎没有氧空位形成,通常用LaMnO3+λ表示,即使用少量Sr进行掺杂,也不形成含氧空位的结构。而Fe系钙钛石则容易形成氧空位,在LaxSr1-xFeO3-λ钙钛石型复合氧化物中,即使没有低价Sr的掺杂,也会存在一定的氧空位[15];通过碱土元素Sr取代三价稀土元素La,可以同时控制B位Fe离子的价态和氧空位数量,通常用下方程表示:La1-xSrxFe3+1-xFe4+xO3→LaFeO3+ x La3+|Sr2+|′+x Fe3+|Fe4+|·或La1-xSrxFe3+(VO)λO3-λ→LaFeO3+x La3+|Sr2+|′+λ|O|··(λ=x/2)。许多文献也强调了氧空位对稳定贵金属纳米粒子有重要的作用[6,16],因此,Pt纳米粒子在Fe系钙钛石载体上可以实现均匀且稳定分散。

图3 不同钙钛石负载Pt催化剂的TEM照片Fig.3 TEM images for various nano-Pt particle and 1%Pt/ABO3
2.4 H2-TPR表征
钙钛石复合氧化物上的H2-TPR曲线上一般有3组还原峰,分别为α(150~450℃),β(450~700℃)和γ(≥700℃)峰。α,β和γ峰分别归属于H2与O2-,与高价金属离子结合的晶格氧和晶格氧生成水,并对应着样品(过渡金属离子)的氧化当量[17]。图4为不同钙钛石及其Pt负载催化剂的H2-TPR谱。从图中可以看到,Sr掺杂后,Mn系钙钛石α还原峰强度略有下降,γ还原峰略向高温方向偏移,说明Sr掺杂后Mn系钙钛石的氧化还原能力略有下降;而50% Sr掺杂后,Fe系钙钛石α,β,γ还原峰强度显著增加,且γ还原峰向低温方向偏移。这也从另一个方面也说明了Mn系钙钛石不含有氧空位。对Fe系钙钛石而言,Sr掺杂后钙钛石表面吸附氧数量增加,晶格氧的可移动性增强,一方面说明钙钛石的氧化还原能力增强,一方面也说明了其氧空位的含量在增加。将Mn系与Fe系进行对比,发现Mn系钙钛石低温的氧化性能要明显强于Fe系,这预示着其有较好的低温的活性。与纯钙钛石相比,1%Pt负载的Mn系钙钛石在109℃处出现了低温还原峰,且Mn系钙钛石样品的H2还原峰都向低温移动了,说明Pt的存在促进了锰系钙钛石的低温还原性能;负载Pt的Fe系钙钛石的还原峰与未负载的相比,变化不明显。这说明Pt负载的锰系钙钛石较铁系钙钛石有更强的氧化还原性能。
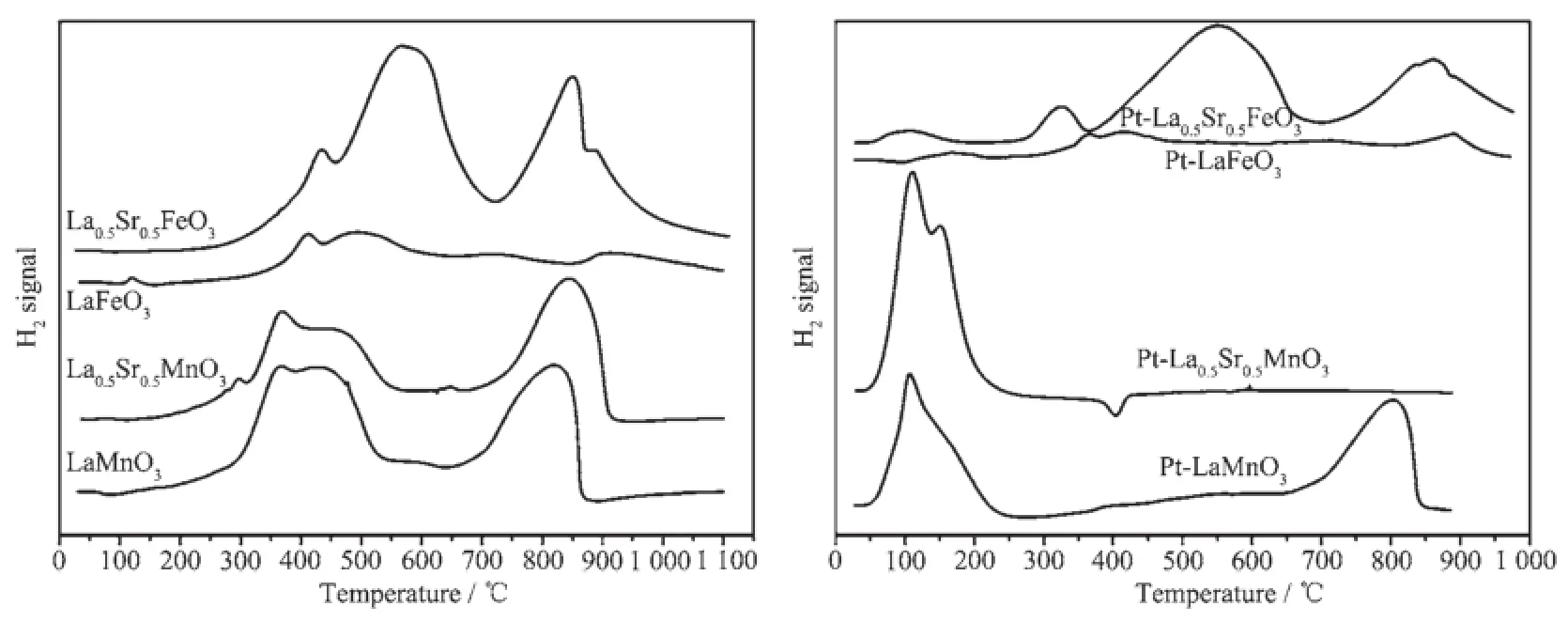
图4 不同钙钛石及其负载Pt催化剂的H2-TPR谱Fig.4 H2-TPR profiles of various ABO3mixed oxdes and 1%Pt/ABO3
对Mn体系,不论是否用Sr进行掺杂,其低温氧化能力与MnO2一样都是非常强的,由于Mn系钙钛石几乎不含氧空位,H2还原过程仅涉及吸附氧和表面晶格氧;而对铁体系,只有在Sr掺杂后,Fe的价态升高,氧空位含量也增加,因而显示出较大的H2-还原峰。引入贵金属Pt后,由于Pt对氢的活化作用进而促进Mn体系还原峰向低温迁移;对铁体系,虽然Pt能够促进H2的活化,但对高价铁的还原影响较小,这也与氧化铁还原能力差相一致。

图5 不同钙钛石及其负载Pt催化剂的CO氧化催化性能Fig.5 Catalytic properties of CO oxidation over various ABO3mixed oxdes and 1%Pt/ABO3
2.5 CO氧化活性
图5给出了不同钙钛石及其负载Pt后的CO氧化性能。对Mn系钙钛石,由于其具有较强的氧化还原性能,因而显示出较高的CO氧化活性,在250℃时CO的转化率达到100%。掺杂Sr后,CO氧化活性几乎没有改进,这与TPR的结果是一致的,低温氧化活性与催化剂的氧化还原性能有关。负载纳米Pt粒子后,CO氧化活性有非常明显的提高,100%CO转化的温度从250℃,降到150温度,同时Sr掺杂的钙钛石明显高于未掺杂的。对Fe系钙钛石,由于其弱的氧化-还原性能,LaFeO3和La0.5Sr0.5FeO3催化剂上CO完全氧化活性非常低,在350℃时CO才能完全转化,其中,LaFeO3的活性略好于La0.5Sr0.5FeO3的,可能与其表面碱性有关,Sr的掺杂会增加催化剂的表面碱性,对CO2有较强的吸附能力(见图2,SrCO3峰)。然而,当将纳米Pt粒子负载在Fe系钙钛石上,CO的氧化活性出现显著变化,CO完全转化的温度从350℃降到120℃,Fe系钙钛石CO氧化的活性明显高于Mn系的,这一结果表明这种显著促进作用源于Fe系具有氧空位。
对Mn钙钛石系,其低温CO氧化活性与其自身的氧化能力有关,在引人Pt之后,虽然对其氧化能力有所改善,但对分子氧吸附活化能力没有得到明显改善,所以Mn系钙钛石加入Pt,催化剂对CO氧化活性的促进作用不够显著;但对铁体系,加入Pt后其氧化能力显著增加,而铁系钙钛石自身又具有吸附活化氧的特性,因而,对CO氧化活性的促进作用就显得非常明显。对铁系钙钛石,没有Sr调变时,其氧空位量对氧的吸附活化能力就可以满足纳米Pt氧化CO的需要,通过Sr调变增加氧空位量,虽进一步增加对氧化的活化能力,此时对CO氧化活性的影显得不再重要。铁系钙钛石的结果表明:吸附CO的Pt与吸附氧的铁钙钛石的结合具有非常强的互补性,因而对CO氧化的活性才会有大幅度改善。
上述结果表明虽然氧化还原性能对氧化反应的作用是至关重要的,但对CO的低温氧化过程还存在着其它因素。如前面所述的,CO氧化性能不仅与催化剂的氧化-还原性能密切相关,也与O2的吸附性能密切相关。
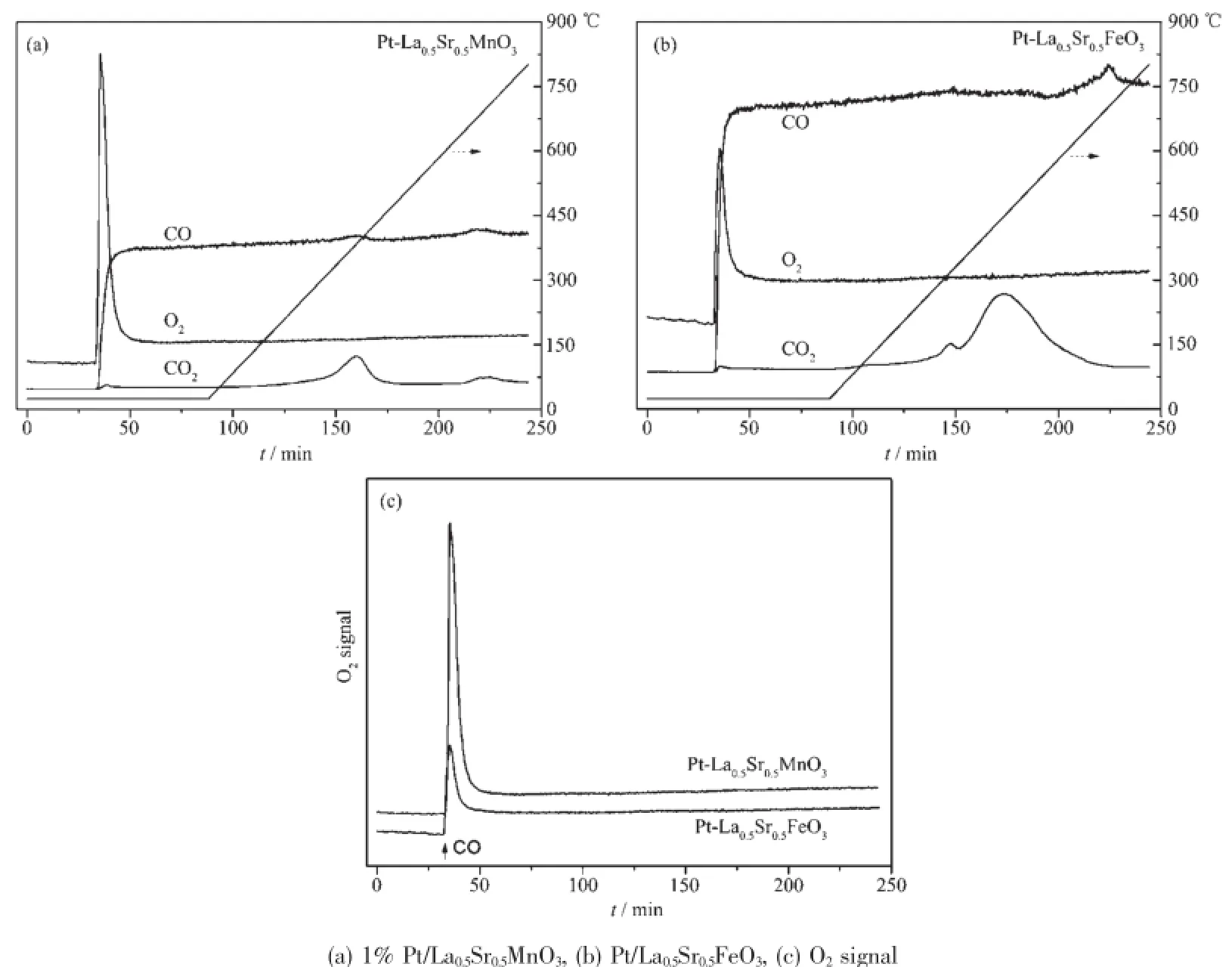
图6 1%Pt负载钙钛石上CO,O2的吸附性能Fig.6 Adsorption properties of CO and O2over 1%Pt/ABO3
2.6 CO、O2的吸附反应性能
在CO+O2反应中,O2的吸附与活化是CO氧化反应过程不可缺少的环节。然而对贵金属催化体系,CO氧化低温活性不高的一个原因是因为CO的强吸附而影响了O2的吸附[1-2,18]。许多文献都给出了~20 kJ·mol-1CO氧化活性能[7],远低于化学吸附活化能40 kJ·mol-1,因此,有理由认为在含贵金属催化体系中,影响CO氧化活性的不是单纯的催化剂对CO活化问题,而是O2与CO或H2O竞争吸附带来的问题。图6给出了负载Pt催化剂上CO,O2的竞争吸附性能和表面反应性能。在预先吸附O2的Pt/La0.5Sr0.5MnO3上,通入1%CO后,出现了较大的O2脱附峰,峰强度明显高于CO的浓度;同时在350℃出现一个较小的CO2脱附峰,表明通入CO后,催化剂表面仅存留较少的表面吸附氧;而对预先吸附O2的Pt/La0.5Sr0.5FeO3,通入CO后,则出现相对较小的O2脱附峰,峰强度明显低于CO的浓度;同时在450℃出现一个较大的CO2脱附峰,表明即使通入1%CO后,表面仍残留大量吸附氧。二者对氧吸附能力差别可以由图6c看出,相对于Pt/ La0.5Sr0.5MnO3,CO能从Pt/La0.5Sr0.5FeO3催化剂上置换出较少的吸附氧,即氧空位对O2的吸附起着重要的作用,这也是影响低温CO氧化活性的一个关键因素。
3 结论
在CO氧化反应中,催化剂的氧化-还原性能对CO的氧化通常起着重要的作用,但对含贵金属体系,会因CO的强吸附抑制了O2的吸附,从而抑制CO氧化活性。对Pt钙钛石体系,因氧空位的存在促进了O2的吸附,因而,而可大幅度提高CO氧化活性。
[1]LIANG Fei-Xue(梁飞雪),ZHU Hua-Qing(朱华青),QIN Zhang-Feng(秦张峰),et al.Prog.Chem.(化学进展),2008, 20(10):1453-1464
[2]Freund H J,Meijer G,Scheffler M,et al.Angew.Chem.Int. Ed.,2011,50(43):10064-10094
[3]Haruta M,Tsubota S,Kobayashi T,et al.J.Catal.,1993,144 (1):175-192
[4]MAO Dong-Sen(毛东森),TAO Li-Hua(陶丽华),WANG Qian(王倩),et al.Chinese J.Inorg.Chem.(无机化学学报), 2010,26(3):447-452
[5]ZHANG Xin(张鑫),XU Bo-Qing(徐柏庆).Acta Chim.Sinica (化学学报),2005,63(1):86-90
[6]Fernandez-Garcia M,Martinez-Arias A,Iglesias-Juez A,et al. Appl.Catal.B,2001,31(1):39-50
[7]Xie X,Li Y,Liu Z,et al.Nature,2009,458(7239):746-749
[8]Leng M,Liu M,Zhang Y,et al.J.Am.Chem.Soc.,2010, 132(48):17084-17087
[9]YU Qiang(余强),GAO Fei(高飞),DONG Lin(董林).Chinese J.Catal.(催化学报),2012,33(8):1245-1256
[10]Green I X,Tang W,Neurock M,et al.Science,2011,333 (6043):736-739
[11]Fu Q,Li W,Yao Y,et al.Science,2010,328(5982):1141-1144
[12]Pena M A,Fierro J L G.Chem.Rev.,2001,101(7):1981-2017
[13]Wang H,Zhao Z,Xu C,et al.Catal.Lett.,2005,102(3):251-256
[14]Shi L,Du F.Mater.Res.Bull.,2007,42(8):1550-1555
[15]Wu Y,Yu T,Dou B,et al.J.Catal.,1989,120:88-107
[16]Ta N,Liu J,Chenna S,et al.J.Am.Chem.Soc.,2012,134 (51):20585-20588
[17]Liu J,Zhao Z,Xu C M,et al.Appl.Catal.B,2008,78(1/2): 61-72
[18]Liu K,Wang A,Zhang T.ACS Catal.,2012,2(6):1165-1178
Action of Oxygen Vacancy in CO Catalytic Oxidation over Pt Supported Pervoshkite Oxides
KONG Ling-Zhi1WAN Qian1XU Li2YAN Yong-Sheng2LIHua-Ming*,2YANG Xiang-Guang*,3
(1School of Energy and Power Engineering,Jiangsu University,Zhenjiang,Jiangsu 212013,China)
(2Institute for Energy Research,Jiangsu University,Zhenjiang,Jiangsu 212013,China)
(3Changchun Institute of Appled Chemistry,Chinese Academy of Sciences,Changchun 130022,China)
LaMnO3,LaFeO3,La0.5Sr0.5MnO3,La0.5Sr0.5FeO3were prepared by citric acid method and Pt/LaMnO3,Pt/ LaFeO3,Pt/La0.5Sr0.5MnO3,Pt/La0.5Sr0.5FeO3were prepared by impregnating nano-Pt solution to ABO3mixed oxides. It showed that prepared ABO3mixed oxides possessed pervoskite structrue from XRD and IR data,the size of nano-Pt particle was about~3 nm and dispersed uniformly on ABO3mixed oxides from TEM.It was found that the redox properties determined the catalytic properties of ABO3-type catalsysts in CO catalytic oxidation,thus, Mn-ABO3catalsysts gave the better activity in CO catalytic oxidation.When supported nano-Pt paricle to ABO3-mixed oxides,Pt/Fe-ABO3catalsysts showed the excellent activity,the reaction temperature of 100%CO conversion was reduced from 350℃to 120℃.Adsorption experiment indicated that oxygen vacancy on ABO3mixed oxides was a key factor to improve the adsorption of oxygen and to determine the properties of CO oxidation at lower reaction temperatures.
perovskite;nano-Pt particle;redox;O2adsorption
O643.3
A
1001-4861(2015)07-1335-07
10.11862/CJIC.2015.163
2015-01-05。收修改稿日期:2015-04-22。
国家自然科学基金(No.21273221)资助项目。
*通讯联系人。E-mail:xgyang@ciac.ac.cn,lihm@ujs.edu.cn,Tel:0511-88791108;会员登记号:S06N566P1004。
