Perl语言整合5款软件进行真菌效应蛋白预测
2015-11-29冯世鹏
冯世鹏
海南省热带生物资源可持续利用重点实验室,海南大学 农学院,海南 海口 570228
依据植物对病原物的抗病机制不同,将植物抗病性分为被动抗性(passive resistance)和主动抗性(active resistance)。被动抗性(也称物理化学抗性)是植物自身性状所决定的抗病性,如植物通过结构屏障(如角质层、蜡质等)和体内的化学成分(如次生代谢产物等)阻止病原物浸入[1]。主动抗性指植物在受到病原物侵染或诱导剂处理时诱导激活一系列防卫反应,抑制病原物浸染,包括一系列生理生化的变化和抗病相关基因的激活表达,如侵染部位的木质化、富含羟脯氨酸的糖蛋白增加、植保素(phytoalexins)积累等[2]。
植物的主动抗病性主要借助于植物的先天免疫反应,可分为2 种。第一种是基于细胞表面的模式识别受体(pattern-recognization receptors,PRR)对病原物相关分子模式(pathogen-associated molecular patterns,PAMP)的识别,该免疫过程被称为PAMP触发的免疫(PAMP triggered immunity,PTI),该免疫过程可帮助植物抵抗大部分病原微生物的入侵;第二种免疫发生在细胞内部,主要依靠抗病基因(resistance gene,R gene)编码的蛋白产物直接或间接地识别病原微生物产生的效应因子(effector)并激发免疫反应,也称为效应因子触发的免疫(effector triggered immunity,ETI)。
广义的效应因子是指病原物可引起宿主细胞结构和功能改变的蛋白或小分子物质,它们促进侵染进程,或激发免疫反应,或两者都有。广义的效应因子包括PAMP、毒素、降解酶等[3],其中蛋白类是效应因子中的重要组成部分,也称为效应蛋白(effector proteins)[4]。不同物种效应蛋白结构有差异,如细菌通过Ⅲ型分泌系统(type Ⅲsecretion system,T3SS)分泌效应蛋白[5-6],卵菌的效应蛋白含有RXLR 结构域[7]等。
真菌效应蛋白的特点是:①无典型的结构域。虽然有些研究发现部分效应蛋白有2 个或多个亮氨酸,以形成如二硫键这类结构,但整体来说,真菌效应蛋白没有共有的结构特征[8],效应蛋白之间及其与其他蛋白间的相似性均不高,因此在进行预测时常与Swiss-prot 数据库进行blast 比对,无显著相似结构蛋白作为潜在效应蛋白的条件之一。②属于胞外分泌蛋白。蛋白分泌至细胞外主要有2 条途径,一是经典途径,通过蛋白质N 端信号肽序列将其引导分泌至胞外,约90%的分泌蛋白通过这种途径分泌至胞外[9];另一条是非经典途径,少数分泌蛋白通过该途径分泌至胞外[10]。在进行效应蛋白预测时经常考虑的是经典途径分泌的蛋白,主要通过蛋白N 端信号肽来预测分泌蛋白。
有多款软件可进行分泌蛋白的预测,每款软件的预测方法及功能各有侧重,Caccia等[11]对这些软件及其作用进行了总结。将各软件按照预测的结果来分类,可分为如下几类:①蛋白信号肽及其剪切位点预测,常见的预测软件如SignalP[12],其他功能类似的软件有PrediSI[13]、SigCleave[11]、SpScan[11]、Phobius[14-15]、Philius[16]、RPSP[17]等;②非经典途径分泌蛋白预测,常见的预测软件如SecretomeP[18],其他功能类似的软件有SOSUI-GramN[19],NClassG+[20]等;③蛋白亚细胞定位预测,常见的如ProtComp[11]、TargetP[21]、WoLF PSORT[22],其他有ChloroP[23]等;④跨膜蛋白预测,常见的预测软件有TMHMM[24]、TMpred[25]。
对分泌蛋白的预测使用一款软件如SignalP[26]或者WoLF PSORT[27],或者联合使用多款软件,多款软件有不同的组合形式[28-33]。
综合已有文献报道的分泌蛋白软件预测使用情况,结合效应蛋白的特征,本研究拟用Perl语言整合SignalP4.1、ProtComp、TargetP1.1、TMHMM2.0、WoLF POSRT等5款软件进行真菌效应蛋白预测。
1 材料和方法
1.1 测试数据来源
香蕉枯萎病菌(Fusarium oxysporumf.sp.cubense race 1)为镰刀菌属的真菌,基因组大小约50 Mb,其蛋白质组包含15438 条蛋白序列(BioProject Accession:PRJNA174274),在NCBI 下载这些蛋白序列进行测试。
1.2 真菌潜在效应蛋白确定
本文将无典型结构域的胞外分泌蛋白定义为潜在的真菌效应蛋白。
真菌效应蛋白无典型结构域,大多数效应蛋白的功能未知。在NCBI中登录的蛋白,其功能有实验验证的,将其实验验证结果列出;暂未进行实验验证的,会通过nr、Swiss-prot、KEGG、interpro、COG 等数据库的blast 搜索,根据相似蛋白的功能来推测未知蛋白的功能(即蛋白注释);其中功能既无实验验证结果又与其他数据库蛋白无显著相似性的蛋白常被命名为“hypothetical protein(预测蛋白,其功能未知)”。根据这种特点,本文拟通过名称中出现的“hypothetical protein”这一名词区分与其他蛋白无显著相似性的蛋白,这样无须进行相关数据库的blast 搜索(蛋白注释过程也完成这一步,因此可省略)。对于研究者自行测序的新物种,先对所预测蛋白进行注释,注释完的数据也可利用本文提供的方法进行后续分析。
真菌效应蛋白为分泌蛋白,这类蛋白含有信号肽序列,定位于细胞外,并非跨膜蛋白。通过软件SignalP 预测蛋白N 端含有信号肽序列,通过3 款亚细胞定位软件ProtComp、TargetP、WoLF PSORT预测蛋白均定位于胞外,通过软件TMHMM 预测不是跨膜蛋白。
1.3 预测软件介绍
信号肽预测软件SignalP4.1。通过神经网络法进行N 端信号肽及其剪切位点预测,包含5 个得分参数,即C值(剪切位点原始分值)、S值(信号肽序列分值)、Y 值(C 值几何均值与S 值的联合得分值)、mean S(S 值的算术均值)、D 值(mean S 值与最大Y值的权重均值)。其中D 值用于区分信号肽(SP=‘YES’)与非信号肽(SP=‘NO’)。本研究使用参数D值“YES”,预测蛋白含有N 端信号肽序列。SignalP是应用最广泛的预测软件之一,Choi 等[26]测试发现其对分泌蛋白的预测准确性达89%,因此可单独用于蛋白分泌组预测。安装软件版本signalp-4.1c.Linux.tar.gz(下载地址:http://www.cbs.dtu.dk/services/SignalP/)。
亚细胞定位预测软件ProtComp。可预测蛋白定位于何种细胞器或区域,如Nuclear(核)、Plasma membrane(质膜)、Extracellular(胞外)、Cytoplasmic(细胞质)、Mitochondrial(线粒体)、Endoplasm.retic.(内质网)、Peroxisomal(过氧化物酶体)、Lysosomal(溶酶体)、Golgi(高尔基体)、Vacuolar(液泡)。该软件使用5 种预测方法,即LocDB(与已知定位蛋白同源比对)、PotLocDB(与理论分析定位蛋白同源比对)、NNets(神经网络法)、Pentamers(查询序列与数据库序列的pentamers 分布比较法)和Integral(将上述4 种方法分值进行最终网络法整合),预测结果供研究者自行判断。本研究选用Integral 值“Extracellular”预测蛋白定位于胞外。安装软件版本protcompan.tar.bz2(下载地址:http://linux1.softberry.com/berry.phtml?topic=index&group=programs&subgroup=proloc)。
亚细胞定位预测软件TargetP1.1。依据N 端的一定序列长度预测真核生物蛋白质定位(Loc)于叶绿体(C,含叶绿体转运肽cTP)、线粒体(M,含线粒体转运肽mTP)、含信号肽分泌蛋白(S,含信号肽序列SP),或者其他地方(-)。通过预测的最高值与第二高值之间的差异分值(diff)进行可信度分区,1 代表diff>0.8,2 代表0.8>diff>0.6,3 代表0.6>diff>0.4,4 代表0.4>diff>0.2,5代表diff<0.2。本研究使用参数Loc值“S”,预测蛋白含信号肽序列并分泌至胞外。安装软件版本targetp-1.1b.Linux.tar.Z(下载地址:http://signalfind.org/tatfind.html)。
亚细胞定位软件WoLF PSORT。可预测蛋白定位于extr(extracellular,胞外)、cysk(cytoskeleton,细胞骨架)、E.R.(endoplasmic reticulum,内质网)、golg(Golgi apparatus,高尔基体)、mito(mitochondria,线粒体)、nucl(nucleus,细胞核)、plas(plasma membrane,细胞膜)、pero(peroxisome,过氧化物酶体)、vacu(vacuolar membrane,液泡膜)、chlo(chloroplast,叶绿体)、lyso(lysozyme,溶酶体),并对可能的定位情况进行打分,分值越高定位于该处的可能性越高。本研究使用extr 值>17,预测蛋白定位于胞外。O'Connell等[27]发现其对胞外分泌蛋白预测准确性达92.7%,因此单独用其进行真菌分泌组蛋白预测。安装软件版本WoLFPSORT_package_v0.2(下载地址:http://wolfpsort.org/)。
跨膜蛋白预测软件TMHMM2.0。预测蛋白跨膜螺旋的数量(Number of predicted TMHs,PredHel)和跨膜区氨基酸长度(Exp number of AAs in TMHs,ExpAA),同时给出查询蛋白长度值(len)、前60 个氨基酸所预测跨膜螺旋数(First60)、及拓扑结构(Topology)等参数。ExpAA 长度大于18 则证明有跨膜结构或者有信号肽。本研究使用参数ExpAA值<18,预测蛋白不是跨膜蛋白。安装软件版本tmhmm-2.0.Linux.tar.gz(下载地址:http://www.cbs.dtu.dk/services/TMHMM/)。
所使用电脑为Dell 工作站T5600,内存32G,操作系统CentOS6.5,系统自带的Perl 版本5.10.1,所有软件的安装根据相关软件说明进行,安装完毕将相关命令输出至环境变量,保证该命令可直接调用。
相关软件及其参数见表1。
1.4 使用Perl语言进行软件整合
1.4.1 方法一:让各软件对蛋白质组进行平行预测再取结果交集 将蛋白质组分为各个蛋白质后,每一款软件都对任一蛋白进行预测,各软件的排列顺序对该方法最终运行时间及最终预测结果均无影响(图1)。用该方法可获得每一款软件对整个蛋白质组完整的预测结果(*.signalp.out、*.protcomp.out、*.targetp.out、*.tmhmm.out、*.wolfpsort.out)及最后的各软件预测结果的交集,即潜在的效应蛋白(*.effector.list、*.effector.fa、*.effector.info),其中*.effector.list 文件存放的是潜在效应蛋白名称清单,*.effector.fa 文件存放fasta格式的潜在效应蛋白序列,*.effector.info文件存放的是5 款软件对各潜在效应蛋白预测结果的汇总。此方法的优点是可获得所有软件预测结果,缺点是运行时间明显延长。
1.4.2 方法二:让各软件按顺序进行预测,边运行边取交集 将蛋白质组分为各个蛋白质后,让各软件按顺序对所取各蛋白进行预测,边预测边取前面各软件预测结果的交集,如果其中任一款预测软件所预测结果不符合参数设定,则终止其后的软件对该蛋白的进一步预测分项,这样可快速获得最终的交集结果(图2),其中*.signalp.final.out、*.protcomp.out、*.targetp.out、*.tmhmm.out、*.wolfpsort.out 这5 个文件存放的是各软件对潜在效应蛋白的预测结果,这些结果与方法一的结果的区别是:前者保存的各软件对整个蛋白质组完整的预测结果,后者保存的是各软件对最终潜在效应蛋白的预测结果。*.effector.list、*.effector.fa、*.effector.info 这3 个文件存放的内容与方法一同名文件存放相同的内容(表2)。在相关软件的顺序安排上,由于各软件运行速度有差异,会导致最终运行时间的差异,因此将运行速度快的软件或方法尽可能排在前面(表2),由于SignalP是预测信号肽蛋白最准确,也是目前预测分泌蛋白使用最多的软件,因此将其排列在除Linux系统命令之外的其他各软件之前,WoLF PSORT 是另一款可单独用于分泌蛋白预测的软件,将其放在最后,这样最终软件排列顺序依次为Linux 系统命令、SignalP、ProtComp、TMHMM、TargetP、WoLF PSORT。
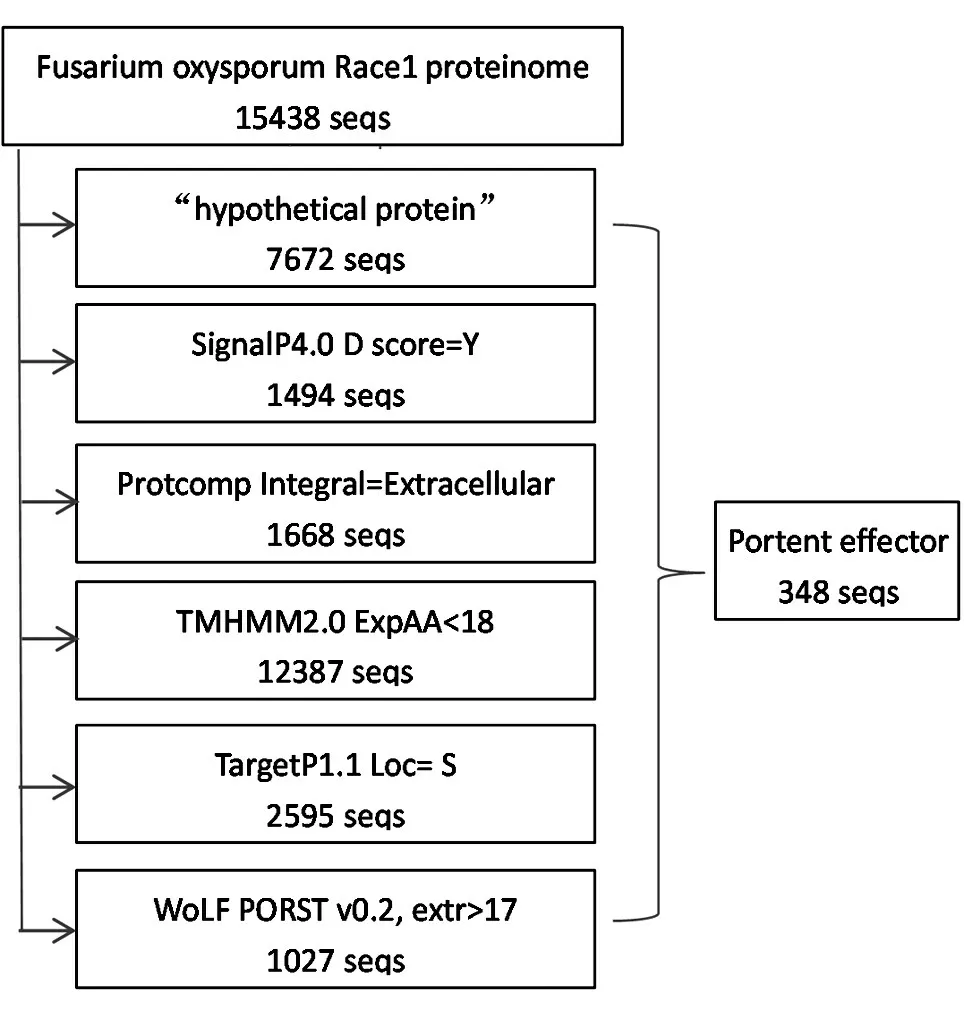
图1 方法一预测流程图(各软件平行预测)
2 结果与讨论
用香蕉枯萎病菌蛋白质组(共有蛋白序列15438 seqs)进行2 种方法的测试,2 种方法均可预测最终潜在效应蛋白348 seqs。
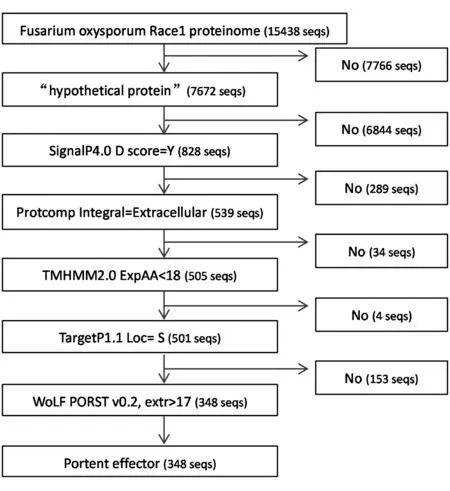
图2 方法二预测流程图(各软件先后预测)
方法一在获得最终潜在效应蛋白的同时,可以获得其他软件单独运行的结果,预测流程及结果见图1,预测过程耗时8 h 25 min。具体结果如下:①用Linux 命令找出“hypothetical protein”7672 seqs;②用SignalP 软件预测含信号肽的蛋白1494 seqs;③用ProtComp 软件预测胞外分泌蛋白1668 seqs;④用TMHMM 软件预测非跨膜蛋白12387 seqs;⑤用TargetP 软件预测胞外蛋白2595 seqs;⑥用WoLF PSORT 软件预测胞外蛋白1027 seqs;⑦各软件预测结果交集即为最终潜在效应蛋白348 seqs。
方法二进行各软件的先后顺序预测,前一款软件预测结果不符合参数设置则不再进行后续软件的预测,对香蕉枯萎病菌蛋白质组15438 seqs 进行预测,流程图见图2,整个过程耗时40 min,相对于方法一大大缩短了预测时间,而所预测的最终潜在效应蛋白一致(348 seqs)。预测过程是这样的:①首先用Linux 命令找出其中的“hypothetical protein”7672 seqs,去掉非“hypothetical protein”7766 seqs;②然后用SignalP 软件预测“hypothetical protein”7672 seqs 中含信号肽序列的蛋白828 seqs,去掉不含信号肽序列的蛋白6844 seqs;③用ProtComp 软件预测828 seqs 中定位于胞外的蛋白539 seqs,去掉定位于其他地方的蛋白289 seqs;④用TMHMM 软件预测539 seqs 中不含跨膜结构的蛋白505 seqs,去掉含跨膜结构域的蛋白34 seq;⑤用TargetP 软件预测505 seqs 中定位与胞外的蛋白501 seqs,去掉定位于其他地方的蛋白4 seqs;⑥最后用WoLF PSORT 软件预测501 seqs 中定位于胞外的蛋白348 seqs,去掉153 seqs,这样获得最终潜在效应蛋白348 seqs。
由于真菌经实验验证的效应蛋白太少,无法进行本方法的准确性测试,但是通过比较现有的蛋白分泌组所使用的方法[28-33],本方法与其类似;O'Connell 等[27]通过blast 比对及单独使用WoLF PSORT 进行2 种炭疽病菌的潜在效应蛋白预测,结果分别为177(禾生炭疽菌,Colletotrichum graminicola)和365 seqs(白菜炭疽菌,C.higginsianum),与本方法的预测结果处于同一数量级。
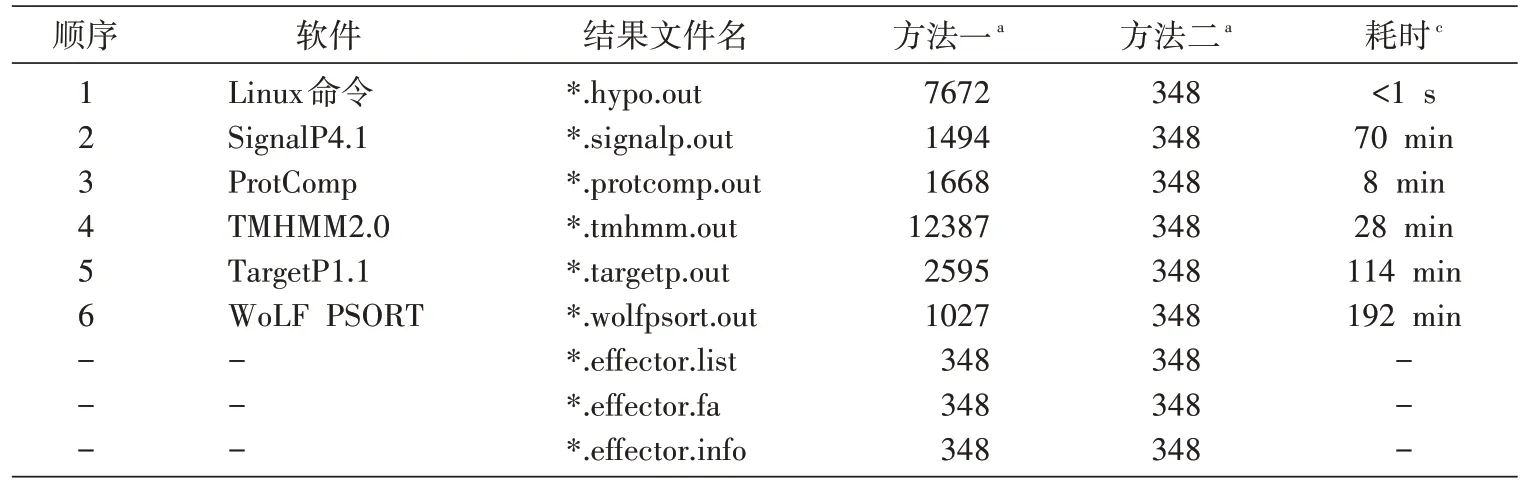
表2 2种方法运行结果比较表
综上,我们整合5 款软件建立了真菌效应蛋白的2 种预测方法,方法一在获得最终潜在效应蛋白的同时,可获得各软件单独的预测结果,但耗时长;方法二可快速获得最终潜在效应蛋白预测结果。2种方法均可用于真菌效应蛋白预测,可由研究者选择使用(2种方法的Perl程序附在文后)。
[1]Hückelhoven R.Transport and secretion in plant-microbe interactions [J].Curr Opin Plant Biol,2007,10(6):573-579.
[2]van Loon L C,Bakker P A,Pieterse C M.Systemic resistance induced by rhizosphere bacteria[J].Annu Rev Phytopathol,2006,36:453-483.
[3]Hogenhout S A,van der Hoorn R A,Terauchi R,et al.Emerging concepts in effector biology of plant associated organisms[J].Mol Plant Microbe Interact,2009,22(2):115-122.
[4]Ellis J,Rafiqi M,Gan P,et al.Recent progress in discovery and functional analysis of effector proteins of fungal and oomycete plant pathogens[J].Curr Opin Plant Biol,2009,12:399-405.
[5]McCann H C,Guttman D S.Evolution of the type III secretion system and its effectors in plant-microbe interactions[J].New Phytol,2008,177(1),33-47.
[6]Block A,Li G,Fu Z Q,et al.Phytopathogen type III effector weaponry and their plant targets[J].Curr Opin Plant Biol,2008,11,396-403.
[7]Dou D,Kale S D,Wang X,et al.RXLR-mediated entry of Phytophthora sojae effector Avr1b into soybean cells does not require pathogen-encoded machinery[J].Plant Cell,2008,20(7),1930-1947.
[8]Houterman P M,Speijer D,Dekker H L,et al.The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants[J].Mol Plant Pathol,2007,8:215-221.
[9]Tsang A,Butler G,Powlowski J,et al.Analytical and computational approaches to define the Aspergillus niger secretome[J].Fungal Genet Biol,2009,46(Suppl1):S153-S160.
[10]Hirai Y,Nelson C M,Yamazaki K,et al.Non-classical export of epimorphin and its adhesion to avintegrin in regulation of epithelial morphogenesis[J].J Cell Sci,2007,120(12):2032-2043.
[11]Caccia D,Dugo M,Callari M,et al.Bioinformatics tools for secretome analysis[J].Biochim Biophys Acta,2013,183(11):2442-2453.
[12]Petersen T N,Brunak S,Heijne G,et al.SignalP 4.0:discriminating signal peptides from transmembrane regions[J].Nat Methods,2011,8:785-786.
[13]Hiller K,Grote A,Scheer M,et al.PrediSi:prediction of signal peptides and their cleavage positions[J].Nucleic Acids Res,2004,32:W375-W379.
[14]Käll L,Krogh A,Sonnhammer E L L.A Combined transmembrane topology and signal peptide prediction method[J].J Mol Biol,2004,338(5):1027-1036.
[15]Käll L,Krogh A,Sonnhammer E L L.Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server[J].Nucleic Acids Res,2007,35:W429-W32.
[16]Reynolds S M,Käll L,Riffle M E,et al.Transmembrane topology and signal peptide prediction using dynamic bayesian networks[J].PLoS Comput Biol,2004,4(11):e1000213.
[17]Plewczynski D,Slabinski L,Ginalski K,et al.Prediction of signal peptides in protein sequences by neural networks[J].Acta Biochim Pol,2008,55(2):261-267.
[18]Bendtsen J D,Jensen L J,Blom N,et al.Feature based prediction of non-classical and leaderless protein secretion[J].Protein Eng Des Sel,2004,17(4):349-356.
[19]Imai K,Asakawa N,Tsuji T,et al.SOSUI-GramN:high performance prediction for sub-cellular localization of proteins in gram-negative bacteria[J].Bioinformation,2008,2(9):417-421.
[20]Restrepo-Montoya D,Pino C,Nino L F,et al.NClassG+:a classifier for non-classically secreted Gram-positive bacterial proteins[J].BMC Bioinformatics,2011,12:21.
[21]Emanuelsson O,Nielsen H,Brunak S,et al.Predicting subcellular localization of proteins based on their N-terminal amino acid sequence[J].J Mol Biol,2000,300(4):1005-1016.
[22]Horton P,Park K J,Obayashi T,et al.WoLF PSORT:protein localization predictor[J].Nucleic Acids Res,2007,35:W585-W587.
[23]Emanuelsson O,Nielsen H,von Heijne G.ChloroP,a neural network-based method for predicting chloroplast transit peptides and their cleavage sites[J].Protein Sci,1999,8(5):978-984.
[24]Sonnhammer E L,von Heijne G,Krogh A.A hidden Markov model for predicting transmembrane helices in protein sequences[J].Proc Int Conf Intell Syst Mol Biol,1998,6:175-182.
[25]Hofmann K,Stoffel W.TMbase -a database of membranespanning proteins segments[J].Biol Chem Hoppe Seyler,1993,374:166.
[26]Choi J,Park J,Kim D,et al.Fungal secretome database:integrated platform for annotation of fungal secretomes[J].BMC Genomics,2010,11:105.
[27]O'Connell R J,Thon M R,Hacquard S,et al.Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses[J].Nat Genet,2012,44(9):1060-1065.
[28]王黎,吉爱国.大肠埃希氏杆菌UTI89 分泌蛋白组的预测分析[J].中国生物工程杂志,2007,27(7):100-105.
[29]周晓罡,丁玉梅,张绍松,等.马铃薯青枯菌(Ralstonia solanacearum)染色体与质粒分泌蛋白组比对分析[J].中国食用菌,2008,27(suppl.):12-15.
[30]李午佼,颜瑾,王运生.南方和北方根结线虫分泌蛋白组预测与比较分析[J].湖南农业大学学报(自然科学版),2011,37(4):376-380.
[31]Lum G,Min X J.FunSecKB:the Fungal Secretome Knowledge Base[J].Database,2011,2011:bar001.
[32]Druzhinina I S,Shelest E,Kubicek C P.Novel traits of Trichoderma predicted through the analysis of its secretome[J].FEMS Microbiol Lett,2012,337(1):1-9.
[33]陈相永,陈捷胤,肖红利,等.植物病原真菌寄生性与分泌蛋白组CAZymes 的比较分析[J].植物病理学报,2014,44(2):163-172.
[34]Emanuelsson O,Nielsen H,Brunak S,et al.Predicting subcellular localization of proteins based on their N-terminal amino acid sequence[J].J Mol Biol,2000,300:1005-1016.
[35]Nielsen H,Engelbrecht J,Brunak S,et al.Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites[J].Protein Eng,1997,10:1-6.
附件1:方法一所用Perl程序

附件2:方法二所用Perl程序

