氨基四氮唑类化合物合成研究进展
2015-11-25王文慧郭冬艳蒋筱莹谢媛媛
王文慧,郭冬艳,蒋筱莹,谢媛媛
(浙江工业大学药学院,浙江杭州310014)
氨基四氮唑类化合物合成研究进展
王文慧,郭冬艳,蒋筱莹,谢媛媛*
(浙江工业大学药学院,浙江杭州310014)
虽然尚未在天然产物中发现,但许多的氨基四氮唑类化合物都具有生物活性,如抗过敏、抗病毒、抗炎、抗肿瘤等活性。综述了氨基四氮唑类化合物的几种有代表性的合成方法,并对合成方法进行了分析总结。
氨基四氮唑类化合物;化学合成;电环化反应
0 前言
杂环化学领域中唑类化合物的合成已经成为最重要的研究课题之一。在各种各样的唑类化合物中,氨基四氮唑类化合物(1)(Figure 1)的合成是一个非常有活力的研究方向。四唑环可作为羧基的生物电子等排体,在改善化合物的生物活性及药代动力学方面起重要作用,被广泛用于药物设计与开发[1]。虽然尚未在天然产物中发现,但许多的氨基四氮唑类衍生物都具有生物活性,如抗过敏[2]、抗病毒[3]、抗炎[4]、抗肿瘤[5]等活性。德国拜耳公司开发的抗HIV药物3’-(5-氨基-1,2,3,4-四唑-1-基)-3’-脱氧胸腺嚓陡核昔及其衍生物(2)[6]如Figure 1所示。

Figure 1
1 合成路线
氨基四氮唑类化合物的合成主要是以睛类、氨基胍类、异硫氰酸醋、硫脲和碳二亚胺类化合物为起始原料,通过与叠氮酸、无机叠氮酸盐或氮化三甲基硅烷的环化反应进行的。下面介绍一些合成氨基四氮唑类化合物的方法。
1.1 氨基胍/NaNO2体系
早在1892年,Kekule[7]等人就以氨基胍为原料经重氮化、异构化合成了氨基四氮唑。1953年Finnegan等人以氨基胍为起始原料经重氮化反应,加热环合生成了氨基四氮唑类化合物。收率87%(Scheme 1)。该工艺步骤繁多,反应条件要求严格。虽然有不少学者对该类工艺提出了改进,但仍存在很多问题。现在已经基本不再使用本法。
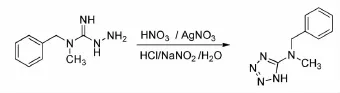
Scheme 1
1.2 碳二亚胺/NaN3体系
1922年Stollé[8]发现了一种合成1-苯基-5-苯胺基四氮唑的方法。他将二苯基碳二亚胺与叠氮化钠在无水乙醇中回流反应5 h(Scheme 2),得到一成环的产物,后证实为1-苯基-5-苯胺基四氮唑。该反应收率并不高。此后又有很多研究者改进了他的方法。

Scheme 2
1979年Svétlík[9]等人研究出一条快速合成1,5-二取代四氮唑的方法,同样也是以碳二亚胺为原料,将HN3与苯的溶液加入反应体系,增加了反应物的活性,因此该反应室温条件下约4min即可反应完毕。收率68%(Scheme 3)。

Scheme 3
叠氮化三甲基硅烷(TMSA)也是一种合成氨基四氮唑的试剂。它与叠氮酸相比,具有相当高的热稳定性,且没有爆炸危险。1980年Tsuge[10]等人报道了一种以TMSA为环化试剂合成氨基四氮唑的方法。将碳二亚胺置于苯与甲醇的混合溶剂中回流20min,收率可达98%(Scheme 4)。该方法虽然产物收率高,但由于溶剂苯毒性较高,现已很少使用。

Scheme 4
反应机理为:叠氮根负离子进攻碳二亚胺的中心碳原子,生成中间体3,三甲基硅烷基连到碳二亚胺的一个氮原子上,叠氮根负离子连到中心碳原子上。然后中间体3经电环化反应生成甲烷硅基化的5-氨基四氮唑4,最后脱甲硅基生成二苯基取代的氨基四氮唑(Scheme 5)。

Scheme 5
1.3 氨腈/NaN3体系
氨睛的结构与碳二亚胺类似,同样可以用来合成氨基四氮唑。采用含有氰基(CN)的化合物和叠氮酸反应是最普遍的合成四氮唑的方法。氯化钱是这类反应常用的无机盐催化剂。
1990年Hantzsch等人报道了用氰胺和氮化钠反应来合成氨基四氮唑的方法。此后又有很多研究者对其进行了改进。
2002年Kanaoka[11]等人以氨睛为原料与NaN3在二甲基甲酞胺中用NH4Cl催化在80℃下反应2.5 h得到产物的收率为66%(Scheme 6)。此反应将NaN3置于NH4Cl中,可防止NaN3爆炸。该方法操作简便、成本低。

Scheme 6
2014年Mehdi[12]等人将上述反应改进,用一种过渡金属的氧化物TiO2替代NH4Cl。二氧化钛无毒,被认为是目前世界上性能最好的一种白色颜料。在110℃下反应1 h,收率达81% (Scheme 7)。

Scheme 7
1.4 Hg盐/NaN3体系
氨基四氮唑类化合物可以直接在HgCl2或Hg(OAc)2的催化下,经电环化反应合成相应的四氮唑。2013年,Ponnuswamy[13]等人报道了一种水介导的三组分一锅法合成氨基四氮唑的方法(Scheme 8)。

Scheme 8
以硫脲为原料,HgCl2促进脱硫反应生成碳二酞亚胺中间体,N3-进攻此中间体参与成环。反应机理见Scheme 9。
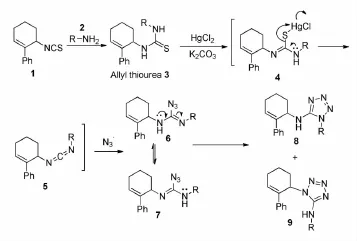
Scheme 9
1.5 高碘试剂
近几年,高碘试剂已经成为有机合成中的一个通用试剂。2012年J.Org.Chem.上的一篇文献[14]报道了一种使用高碘试剂2-碘酞苯甲酸(IBX) (Figure 2)氧化脱硫合成氨基四氮唑的方法。该反应在室温下即可发生,3 h反应完毕,收率90% (Scheme 10)。

Figure 2

Scheme 10
2013年,Telvekar[15]等人又报道了一种以硫脲为原料,在碘苯及氧化剂过硫酸氢钾存在下与叠氮化钠经环化反应生成氨基四氮唑的方法。室温条件下反应3.5 h即可得到88%的收率(Scheme 11)。

Scheme 11
高碘试剂参与的四氮唑类化合物的合成,避免了重金属的使用,反应条件温和,收率高,适用性好。这也为氨基四氮唑类化合物的合成提供了新思路。
2 小结
综上所知,氨基四唑类化合物的合成关键在于碳二亚胺中间体的构建。氨基四唑类化合物作为药物在抗病毒、抗菌、镇痛消炎、抗癌等领域得到了广泛的研究,显示出很高的应用价值。因而开发出更加高效、温和、环境友好的新合成方法具有重要的意义。因此,我们着力于氨基四氮唑类化合物的合成新方法的研究。
[1]代玲玲,崔胜峰,周成合,等.四唑类化合物的合成及应用研究新进展[J].有机化学,2013,33(2):224-244.
[2]Peet N P,Baugh L E,Sundler S,et al.3-(1H-tetrazol-5-yl)-4(3H)-quinazolinone sodium salt(MDL 427):a new antiallergic agent[J].J.Med.Chem,1986,29:2403-2409.
[3]Poonian M S,Nowoswiat E F,Blount JF,Kramer,et al. Synthesis of tetrazole ribonucleosides and their evaluation as antiviral agents[J].J.Med.Chem.,1976,19:1017-1020.
[4]Navidpour L,Shadnia H,Shafaroodi H,et al.Design,synthesis,and biological evaluation of substituted 2-alkylthio-1,5-diarylimidazoles as selective COX-2 inhibitors[J].Bioorg.Med.Chem.,2007,15:1976-1982.
[5]Taveras A G,Mallams A K,Afonso A.Tricyclic inhibitors of farnesyl protein transferase:WO,9811093[P].1998-03-19.
[6]Habich D.Synthesis of 3’-(5-amino-1,2,3,4-tetrazol-1-yl)-3’-deoxythymidines[J].Synthesis,1992,4:358-360.
[7]Kekule A,Erlenmeyer E,Volhard J.Ueber nitro-und am idoguanidin[J].Justus Liebig's Annalen der Chemie,1892, 270:1~61.
[8]StolléR.U ber die Anlagerung von Stickstoffwasserstoffsäure an Carbodiim id-Abkömm linge.(Vorläufige Mitteilung)[J].Chem.Ber.,1922,55:1289-1297.
[9]Světlík J,MartvonňA,Leško J.Preparation and spectral properties of tetrazoles[J].Chem.Zvesti.,1979,33,4: 521-527.
[10]Tsuge Y,Satoshi O U,Oe K.Reactions of trimethylsilyl azide with heterocumulenes[J].J.Org.Chem.,1980,45: 5130-5136.
[11]Yamazaki K,Hasegawa H,Umekawa K,etal.Design,synthesis and biological activity of novel non-peptidyl endothelin converting enzyme inhibitors,1-phenyl-tetrazoleformazan analogues[J].Bioorg.Med.Chem.Lett.,2002,12: 1275-1278.
[12]Mehdi M,Mehdi K.Synthesis of thiotetrazoles and arylaminotetrazoles using rutile TiO2nanoparticles as a heterogeneous and reusable catalyst[J].J.Chem.Res.,2014, 38:502-506.
[13]Sathishkumar M,Shanmugavelan P,Nagarajan S,et al. Water promoted one pot three-component synthesis of tetrazoles[J].New J.Chem.,2013,37,488-493.
[14]Chaudhari P S,Pathare S P,Akamanchi K G.o-Iodoxybenzoic acid mediated oxidative desulfurization initiated dom ino reactions for synthesis of azoles[J].J.Org.Chem., 2012,77:3716-3723.
[15]Jadhav N C,Jagadhane PB,Patel K N,et al.An expedient route to the azoles through oxidative desulfurization using iodine reagent[J].Tetrahedron Lett.,2013,54:101-105.
The Synthesis Development of Am inotetrazoles
GUO Dong-yan,JIANG Xiao-ying,XIE Yuan-yuan
(College of Pharmaceutical Sciences,Zhejiang University of Technology, Hangzhou,Zhejiang 310014,China)
Although tetrazole and its derivatives rarely occur in nature,the vastmajority of all show the biological activity.5-Aminotetrazoles show anti-allergic and anti-asthmatic,antiviral and anti-inflammatory, antineoplastic activities.This article summarized some representative syntheticmethods of the aminotetrazoles and reviewed the advantages and disadvantages of thesemethods.
aminotetrazoles;chemical synthesis;electrocyclization
1006-4184(2015)9-0015-04
2015-02-13
王文慧(1993-),男,本科在读。研究方向:药物中间体合成。E-mail:975102519@qq.com。
*通讯作者:谢媛媛,E-mail:xyycz@zjut.edu.cn。
