井冈霉烯胺的合成路线研究
2015-11-23王红英姜国平浙江迪耳化工有限公司浙江金华321016
王红英,姜国平(浙江迪耳化工有限公司,浙江金华321016)
生物化工
井冈霉烯胺的合成路线研究
王红英,姜国平
(浙江迪耳化工有限公司,浙江金华321016)
综述了国内外以单糖衍生物、肌醇甲醚、奎宁酸、L-酒石酸和其他非糖类化合物为原料合成井岗霉烯胺的方法和路线。
井岗霉烯胺;2,3,4,6-四-O-苄基-D-葡萄糖;D-木糖;肌醇甲醚;奎宁酸;L-酒石酸
1 概述
井冈霉烯胺是从吸水链霉菌limoneus IFO12703的发酵液中分离出来的具有生物活性的假糖胺类化合物,其自身对α-D-葡萄糖苷水解酶具有特异性抑制[1],并有一定的抗菌活性[2]。井冈霉烯胺是阿卡波糖的前体[3]、有效霉素[4]的结构单元,且其结构中的不饱和的氨基环醇可转变为饱和的氨基环醇,N-取代的饱和氨基环醇衍生物伏格列波糖是假低聚糖苷酶抑制剂[5]。
对井冈霉烯胺的化学合成研究始于上世纪八十年代,在近四十年的探索中,国内外研究者分别以单糖衍生物和非糖类物质为原料,采用保护、氧化、还原、重排、脱除保护等方法,生成侧链氨基环己烯化合物井冈霉烯胺。本文分析和整理出所有涉及的合成方法,分别叙述了以单糖衍生物、肌醇甲醚、奎宁酸、L-酒石酸和其他非糖类化合物为原料合成井岗霉烯胺的方法和路线。
2 合成路线
2.1 单糖衍生物
2.1.1 甲基吡喃葡萄糖苷
Richard R.Schmidt等[6]将甲基吡喃葡萄糖苷的6位碘化、2、3、4位乙酰化,脱碘化氢,脱乙酰、苄基化后二价银盐催化重排得到环己酮衍生物,酮转化成酮缩硫醇并继续与三甲基氰硅烷反应生成硫氰,氰还原为醛并苯甲酰化,羟基所在的位置脱水,用氯胺T消除硫醚基、引入氨基,脱去保护得到井岗霉烯胺。如图1所示。
2.1.2 四苄基葡萄糖
杨光丽等[7]将2,3,4,6-四苄基葡萄糖氧化成糖酸1,5内酯,然后开环、闭环生成肌醇单酮,再异构化为环己烯酮,环己烯酮还原为环己烯醇后引入氨基侧链,脱去苄基保护得到井岗霉烯胺。如图2所示。
Young-Kil Chang等[8]先使2,3,4,6-四苄基葡萄糖开环、用乙酸酐和二甲基亚砜氧化,然后脱氧、烯丙基化、烯烃易位环合成环己烯,环己烯再经叠氮和胺化引入氨基,脱去苄基保护得到井岗霉烯胺。如图3所示。
2.1.3 D-呋喃葡萄糖衍生物
Masayuki Yoshikawa等[9]以呋喃葡萄糖为原料合成井岗霉烯胺。双丙酮葡萄糖3位氧化,还原后苄基化,脱去5,6位异丙基保护(或以3-O-苄基-1,2-O-亚异丙基葡萄糖为原料)并苯甲酰化6位,氧化并硝甲基化5位[10],脱去1,2位保护,与氟化钾反应转化成硝基环醇并乙酰化,硝基进行N-乙酰化,消除硝基,脱水,脱去酰基、苄基和N-乙酰基保护得到井岗霉烯胺。如图4所示。

图1

图2

图3
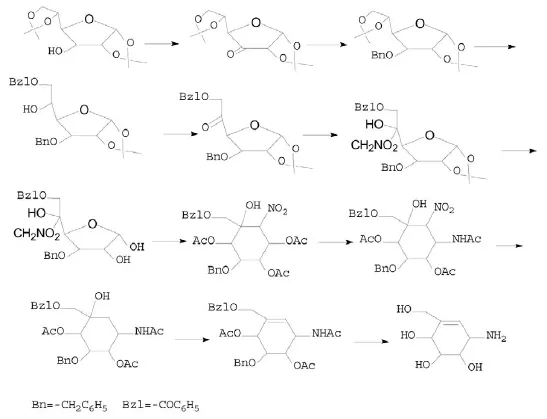
图4
2.1.4 D-木糖衍生物
Kuniaki Tatsuta等[11]将D-木糖的1位氧化、5位三苯甲基化,2位和3位四丁基硅烷基化并脱去三苯基,Pfitzner-Moffatt氧化并甲苯磺酰化,开环,闭环形成环己烯酮,再经Michael反应引入羟甲基,还原羰基,羟基保护并脱除四丁硅烷基,Mitsunobu反转、叠氮化,叠氮基还原为氨基,脱除保护得到井岗霉烯胺。如图5所示。

图5
2.2 肌醇甲醚
Hans Paulsen和Fred R.Heiker[12]以肌醇甲醚为原料,将其转换成二亚异丙基化合物,催化氧化成酮,立体选择性环氧化,水解环氧环,断裂两个二甲醚键并解块,三异丙基化,选择性水解反式亚异丙基并苄基化,然后再水解剩余的两个亚异丙基,经选择性苯甲酰化、甲磺酰化、环氧化、乙酰化、消除环氧环形成环己烯酮,脱除酰基,选择性苯甲酰化,羟基叠氮化,叠氮基还原为氨基并脱去保护得到井冈霉烯胺。如图6所示。

图6
2.3 奎宁酸和其他非糖化合物
2.3.1 奎宁酸
Stanton H.-L.Kok等[13]将奎宁酸经6步反应得到二醇,其羟基选择性苄基化,脱去另一个羟基,水解亚环己基,与亚硫酰氯反应生成环亚硫酸酯,经配向型SN2反应引入叠氮基,甲磺酰化,通过乙酰化转化构型,脱去乙酰基,叠氮基还原为氨基,脱去保护基得到井岗霉烯胺。如图7所示。
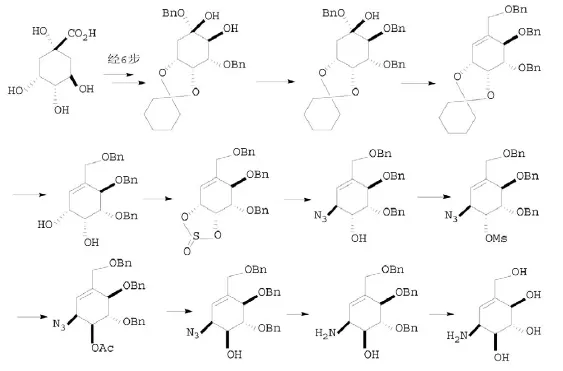
2.3.2 酒石酸
Yuan-Kang Chang等[14]以L-酒石酸为原料合成井岗霉烯胺。首先将其2,3位缩丙酮化保护,然后还原为醇并原位高度非对映选择性添加二乙烯基,易位环合,区域和立体选择性控制环氧化,链迁移重排引入叠氮基,戴斯-马丁氧化剂氧化,经Baylis-Hillman反应引入羟甲基,还原羰基,叠氮基还原为氨基并脱去保护得到井岗霉烯胺。如图8所示。

图8
2.3.3 其他非糖化合物
Barry M.Trost等[15]通过丙炔酸乙酯与带有保护基的间丁二烯衍生物间的狄尔斯-阿尔德反应生成环己二烯,用间氯过氧苯甲酸立体选择性环氧化富电子双键,在碱性催化剂中经E2消除、打开环氧环后甲烷硅基化,继续用间氯过氧苯甲酸环氧化,在保留原构型的基础上添加异氰酸酯,醇解,还原羰基,脱除甲苯磺酰基和甲烷硅基得到井岗霉烯胺。如图9所示。此外,近年来Palakodety Radha Krishna[16]和Bing Zhou等[17]分别以L-丝氨酸和Garner醛为原料合成井岗霉烯胺。

图9
3 结束语
井冈酶烯胺是有效霉素和人体降糖药物伏格列波糖和阿卡波糖的重要中间体,探寻到原料经济、来源广泛,工序相对简单、易操作、收率高及环境污染小的合成工艺将大大降低上述降糖药物的成本,并促进国内降糖药物的生产和开发。
[1]Kameda Y,Asano N,Yoshikawa M,et al.Valiolamine,a new α-glucosidase inhibiting aminocyclitol produced by streptomyces hygroscopicus[J].J.Antibiot.1984,37:1301-1307.
[2]Kameda Y,Asano N,Yoshikawa M,et al.Valienamine as an α-glucosidase inhibitor[J].J.Antibiotics 1980,33: 1575-1576.
[3]Arakawa K,Simeon G B,Benjamin M,et al.Biosynthetic studies on the a-glucosidase inhibitor acarbose:the chemical synthesis of isotopically labeled 2-epi-5-epi-valiolone analogs[J].Carbohydrate Research 2003,338:2075-2082.
[4]Mahmud T,Xu J,Young U C.Synthesis of 5-epi-[6-2 H2]Valiolone and Stereospecifically Monodeuterated 5-epi-Valiolones:Exploring the Steric Course of 5-epi-Valiolone Dehydratase in Validamycin A Biosynthesis[J].J. Org.Chem.2001,66(15):5066-5073.
[5]Hiroshi F,Satoshi H.Synthesis of a Branched-Chain Inosose Derivative,a Versatile Synthon of N-Substituted Valiolamine Derivatives from D-Glucose[J].J.Org.Chem. 1992,57(13):3642-3650.
[6]Richard R,Schmidt,Arnim K.Synthesis of Valienamine[J]. Angew.Chem.Int.Ed.Eng.1987,26(5):482-483.
[7]杨光丽,刘侠,张硕,杨劲松.2,3,4,6-四-O-苄基井冈霉烯胺的合成[J].化学研究与应用,2009,21(5):730-733.
[8]Chang Y K,Lee B Y,Kim D J,et al.An Efficient Synthesis of Valienamine via Ring-Closing Metathesis[J].J.Org. Chem.2005,70(8):3299-3302.
[9]Masayuki Y,Bae C C,Yoshihiko O,et al.Synthese of validamine,epi-validamine,and valienamine,three optically active pseudo-amino-sugars from-glucose[J].Chem.Pharm. Bull.1988,36(10):4236-4239.
[10]Masayuki Y,Yoshihiko O,Bae C C,et al.Synthesis of(-)-Aristeromycin from D-glucose[J].Chem.Pharm.Bull.1989, 37(9):2555-2557.
[11]Kuniaki T,Hiroshi M,Masaaki T.Novel Synthesis of Natural Pseudo-aminosugars,(+)-Valienamine and(+)-Validamine[J].J.Antibiot.2000,53(4):430-435.
[12]Hans P,Fred R H.Synthese von enantiomerenreinem Valienamin aus Quebrachit[J].Liebigs Ann.Chem.1981:2180 -2203.
[13]Stanton H L K,Lee C C,Tony K M S.A New Synthesis of Valienamine[J].J.Org.Chem.2001,66(21):7184-7190.
[14]Chang Y K,Lo H J,Yan T H.A Flexible Strategy Based on a C2-Symmetric Pool of Chiral Substrates:Concise Synthesis of(+)-Valienamine,Key Intermediate of(+)-Pan cratistatin,and Conduramines A-1 and E[J].Org.Lett. 2009,11(19):4278-4281.
[15]Barry M.T,Louis S.C,Thomas L.Total Synthesis of(±)-and(+)-Valienamine via a Strategy Derived from New Palladium-Catalyzed Reactions[J].J.Am.Chem.Soc.1998, 120(8):1732-1740.
[16]Palakodety R K.Stereoselective Total Synthesis of(+)-Valienamine and(+)-4-epi-Valienamine via a Ring-Closing Enyne Metathesis Protocol[J].Synlett 2009(2): 209-212.
[17]Zhou B,Luo Z,Lin S,et al.A Concise Synthetic Approach to(+)-Valienamine Starting from Garner's Aldehyde[J]. Synlett 2012(6):913-916.
Research for Synthetic Route of Valienamine
WANG Hong-ying,JIANG Guo-ping
(Zhejiang Deyer Chemical Co.,Ltd.,Jinhua,Zhejiang 321016,China)
This paper summarises the synthesis of valienamine using monosaccharides derivative,quebrachitol,quininic acid,L-tartaric acid and other non-saccharides compound.
valienamine;2,3,4,6-tetra-O-benzyl-D-glucopyranose;D-xylose;quebrachitol;quininic acid; L-tartaric acid.
1006-4184(2015)3-0030-06
2015-01-08
王红英(1969-),女,工程师,硕士,主要从事药物中间体、塑料助剂等的研究开发工作。E-mail:why@Deyerchem.com。
