新型Atorvastatin类HMGR抑制剂的3D-QSAR和分子对接
2015-11-18蔡明峰叶良春刘卓裕程利平
王 志, 蔡明峰, 叶良春, 刘卓裕, 程利平
(上海应用技术学院化学与环境工程学院,上海 201418)
新型Atorvastatin类HMGR抑制剂的3D-QSAR和分子对接
王 志, 蔡明峰, 叶良春, 刘卓裕, 程利平
(上海应用技术学院化学与环境工程学院,上海 201418)
羟甲基戊二酰辅酶A还原酶(H MGR)是内源性胆固醇合成的重要催化限速酶,通过抑制HMGR的活性可以降低内源性胆固醇的合成.HMGR抑制剂(他汀类)是治疗心血管疾病的最佳药物之一.运用计算机辅助药物设计(CADD)方法,综合三维定量构效关系(3D-QSAR)和分子对接分析研究已合成的H MGR抑制剂的结构与抑制活性之间的关系.根据最优3D-QSAR模型,再结合分子对接分析,设计出新型活性高的H MGR抑制剂分子.
他汀类药物;三维定量构效关系;分子对接;羟甲基戊二酰辅酶A还原酶
近年来,心血管疾病,如冠心病和动脉粥样硬化等,严重威胁着人类健康[1-2],人体内胆固醇过量是心血管疾病产生的主要原因.人体内2/3的胆固醇都是自身合成的,称为内源性胆固醇.羟甲基戊二酰辅酶A还原酶(H MGR)是内源性胆固醇合成第一步的催化限速酶,抑制其催化活性就可以减少内源性胆固醇的合成.HMGR抑制剂(即他汀类药物)是治疗心血管疾病的首选药物之一,自20世纪80年代洛伐他汀(Lovastatin)成功上市,目前已有6种他汀类药物造福心血管疾病患者,阿托伐他汀(Atorvastatin,见图1A)是最畅销的他汀类药物,22号化合物(见图1B)是本文中所采用的Atorvastatin类似物中抑制活性最好的分子.虽然他汀类药物的安全性很高,但是横纹肌溶解的毒性却是其不可避免的副作用,德国拜耳公司的西立伐他汀因横纹肌溶解的副作用而黯然撤市[3],故药物安全性是药物研发最重要的指标.从头设计和传统药物的研发耗时耗材,而基于计算机技术的药物设计对已上市的成熟药物进行合理的结构修饰,得到高活性、低毒的新的分子结构越来越受到药物研发者的欢迎.
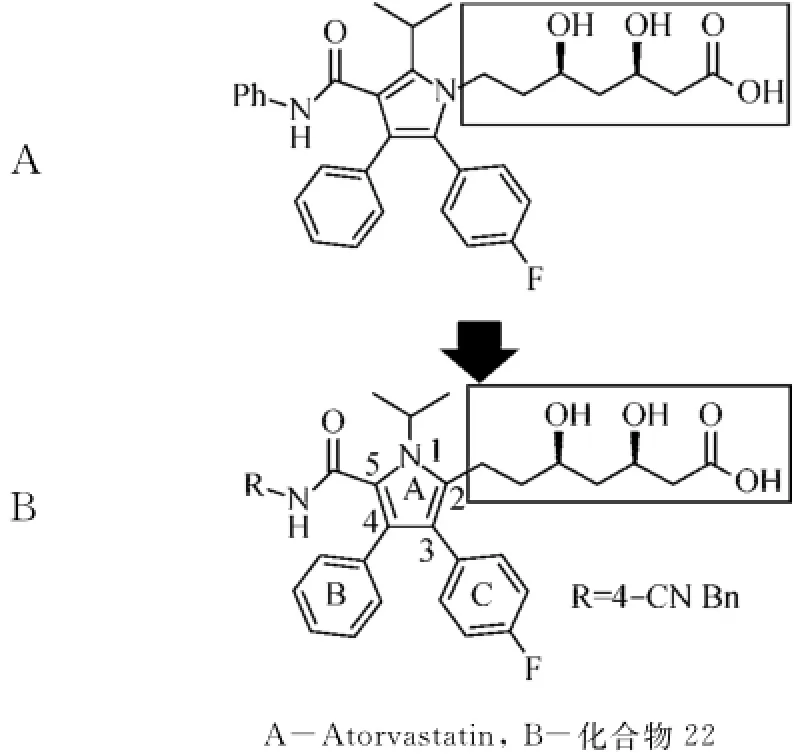
图1 阿托伐他汀和化合物22的结构Fig.1 Structure of Atorvastatin and compound 22
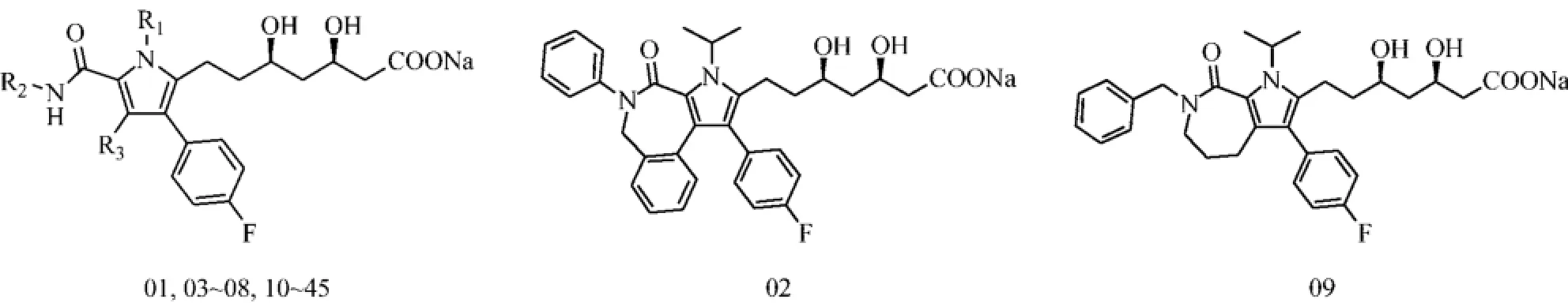
图2 45个分子的结构式Fig.2 Structure of 45 compounds
三维定量构效关系(3D-QSAR)和分子对接是药物设计中常用的方法[4-5],比较分子场分析(Co MFA)和比较分子相似性分析(Co MSIA)是3DQSAR中常用的工具.来源于同一实验室的45个新型Atorvastatin类似物首次被用来建立3D-QSAR模型,抑制活性最好的分子与H MGR蛋白三维晶体进行分子对接研究.综合最优的3D-QSAR模型和分子对接结果,可以设计出抑制活性更高、毒副作用更小的新型他汀类分子.
1 实验部分
1.1 数据集和生物活性
45个Atorvastatin类新分子结构和抑制活性(IC50)由Pfefferkorn[6-8]等合成并发布在期刊上,化合物的IC50值需要转换成p IC50(-log IC50)用作建立3D-QSAR的自变量.将45个分子(结构式见图2)分为2个集合:训练集(35个,77.8%,建立最优模型和内部验证)和测试集(10个,22.2%,外部验证),测试集中与训练集的活性分布相同(见表1). 45个分子在SYBYL-X2.1软件中构建,在Tripos[9]力场,Gasteiger-Huckel[10]电荷和收敛值设为20.93 J/(mol·A),优化次数设定为104,其余参数是在默认设置条件下将所有的分子结构进行能量优化至稳定构象.
1.2 分子建模
分子建模有2种类型:基于配体和基于受体,因基于受体所建立的模型结果较差,故本文只选用了基于配体方式进行分子建模.首先将准备好的训练集数据库在SYBYL软件中打开,运用Align Database[11]模块进行分子叠合工作,经过不同公共骨架的选取,最终选用图3中A部分的结构作为叠合公共骨架建立了最优的3D-QSAR模型.偏最小二乘法(PLS)被用于定量探讨结构参数(CoMFA/CoMSIA相互作用的力场)与生物活性之间的关系,Co MFA中有立体场和静电场2个描述符,Co MSIA中除了有立体场和静电场外,还有疏水场、氢键供体场和氢键受体场3个描述符.在用PLS进行回归分析中,为了评估模型的可靠性,先用抽一法(LOO)中的交叉验证分析生成最佳主成分值(ONC)和交叉验证相关性系数(Q2),再根据最佳主成分值进行非交叉验证得到非交叉验证相关系数(R2)、F统计值和标准估计误差(SEE)来评价模型.当Q2>0.5时,模型是可靠的,通过测试集可以检验模型的预测能力.

图3 公共骨架(A)和叠合模型图(E)Fig.3 The common substructure(A)and alignment model(E)
1.3 分子对接
为了研究他汀类药物分子与HMGR蛋白的相互结合模式,SYBYL-X2.1中的“surflex-docking”模块被用于探索他汀类药物分子对接进入H MGR结合位点的结合机制.首先,从蛋白质数据库(ww w.rcsb.org/pdb/home/home.do,RCSB)中下载H MGR(PDB ID:2Q1L)[12]晶体结构;然后在分子对接前,需对HMGR晶体的结合口袋进行必要的处理,删除晶体中所有水分子和其他非配体的结构,并根据原有配体计算出一个protomol空腔,将结合口袋附近的氨基酸残基去质子化,使得氨基酸残基侧链与抑制剂分子形成更多的氢键和其他较强的相互作用.
2 结果与讨论
2.1 3D-QSAR统计性结果
2.1.1 Co MFA/Co MSIA模型的分析
根据最优模型得到的预测值与实际值见表1,通过计算得到最优的3D-QSAR模型具有良好的统计关系(见表2).预测值与实验值的相关性分析如图4所示,由图可知,预测值与实际值相差不大,说明文中所建模型是可靠的.最优Co MFA模型中,当ONC=5时,Q2=0.546,R2=0.974,SEE=0.091,F统计值为221.039.立体场的贡献值为47.8%,静电场的贡献值为52.2%.说明在CoMFA中,静电场的影响比较大.最优Co MSIA模型中,当ONC= 5时,Q2=0.564,R2=0.965,SEE=0.106,F统计值为162.125.立体场、静电场、疏水场、氢键供体场、氢键受体场的贡献值分别为7.90%、33.3%、 15.5%、21.2%、22.1%.说明在Co MSIA中,氢键场和静电场的贡献较大.
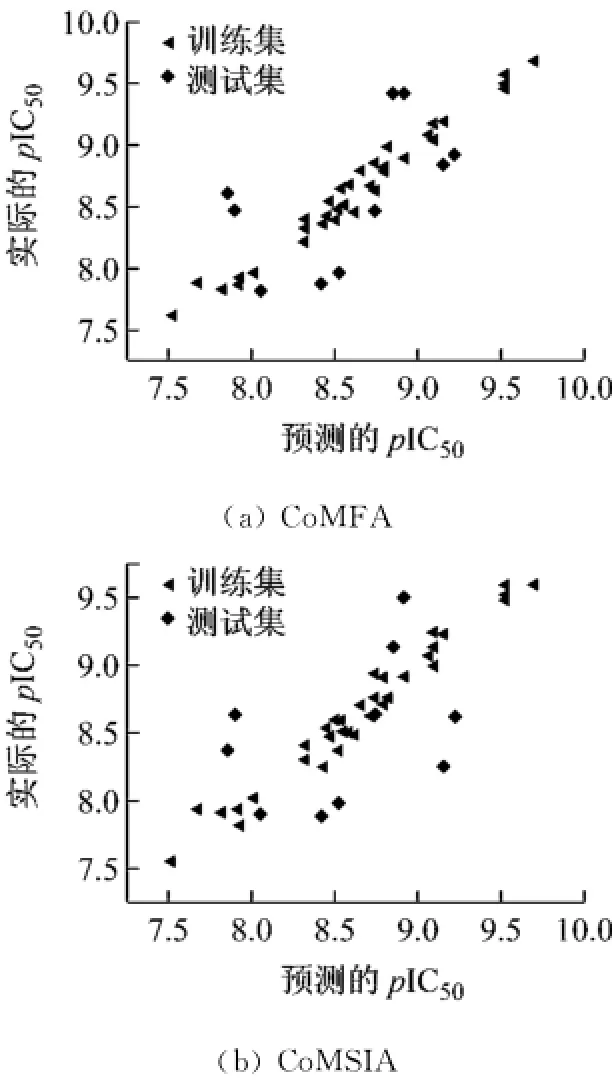
图4 实际和预测p IC50的相关性图Fig.4 The correlation plots of the predicted and the actual p IC50
2.1.2 3D-QSAR等值线图分析
根据最优的3D-QSAR模型,生成了不同力场的等值图,以22号分子作为模板分子(见图1B部分).Co MFA模型生成的等值图如图5所示,图5(a)是静电场的等值图(红色色块表示增加带负电基团会提高活性,蓝色表示带正电基团有利于活性).由图可知,2个中等大小的蓝色色块图位于脂肪酸的末端,表示此处修饰为正电性的原子或基团可以提高活性;除此之外,2个大的蓝色色块分别位于A环的5位和B环的3位,说明正电性的原子或基团对活性有利.2个红色色块环绕着A环5位上的苯环,说明负电性的基团或原子有利于提高抑制剂的活性,如电负性最大的氟原子或含氟基团取代该位置上的原有基团,可以提高抑制剂的活性.图5(b)是立体场的等值图(黄色表示增大基团活性会减弱,绿色表示增大基团活性会提高),1个大的黄色色块环绕着A环4,5位和B环,表示体积小的取代基有利于抑制活性,还有1个中型的黄色色块位于C环的2,3位.1个绿色的小色块位于A环5位上的芳香环的2位,表示此位置取代为体积大的基团可以提高活性.
图6(a)、(b)是Co MSIA模型生成的静电场和立体场的等值图,其模型生成的色块位置与图5基本类似.Co MSIA模型生成的疏水场和氢键场的等值图分别如图6(c)、(d)所示.疏水场的等值图中,白色表示增加亲水性基团有利于活性,黄色表示增加疏水性基团可以提高活性.由图可知,2个大的白色色块分别位于脂肪链和A环5位上芳香环位置. 1个中型的黄色色块位于A环5位上的酰胺基位置,表示这个位置有疏水性基团可以提高活性,因为A环5位连接的基团是疏水的,所以此位置的取代基都是疏水的.氢键场的等值图中,红色表示增加氢键供体不利于活性,紫红色表示减少氢键供体有利于活性;蓝绿色表示增加氢键供体有利于活性,紫色表示减少氢键受体有利于活性.1个小的红色色块和1个大的红色色块分别位于脂肪链的3位羟基和A环5位上酰胺基中的羰基位置,说明此位置的基团是氢键供体的取代基,是高抑制活性的抑制剂分子;1个大的紫红色色块位于A环5位的大的取代基位置,说明此处减少氢键供体有利于活性;2个小的蓝绿色色块分别位于脂肪链的羧酸位置和B环的2位,说明此处增加氢键供体有利于活性;还有A环5位的取代基附近有1个中等大小的蓝绿色色块,1个小的紫色色块位于A环5位取代基上的苯环的2位,表示此位置减少氢键供体有利于活性.综上所述,可以根据最优3D-QSAR模型的等值图设计高活性的化合物.
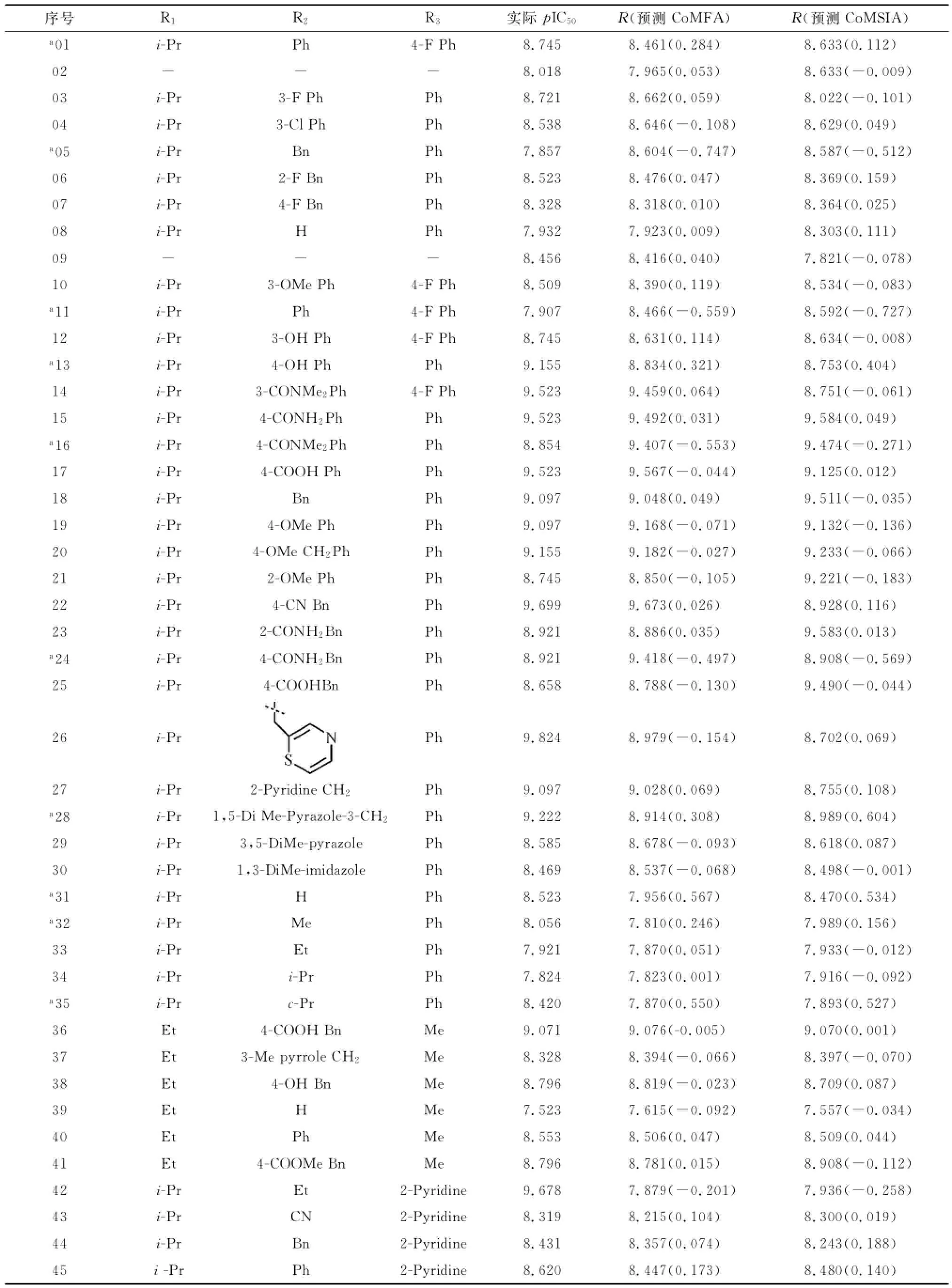
表1 抑制剂分子结构和p IC50的实际值与预测值Tab.1 The structures of inhibitor molecules and the actual and predicted p IC50

表2 Co MFA和Co MSIA模型的统计结果Tab.2 Statistical results of Co MFA and Co MSIA models
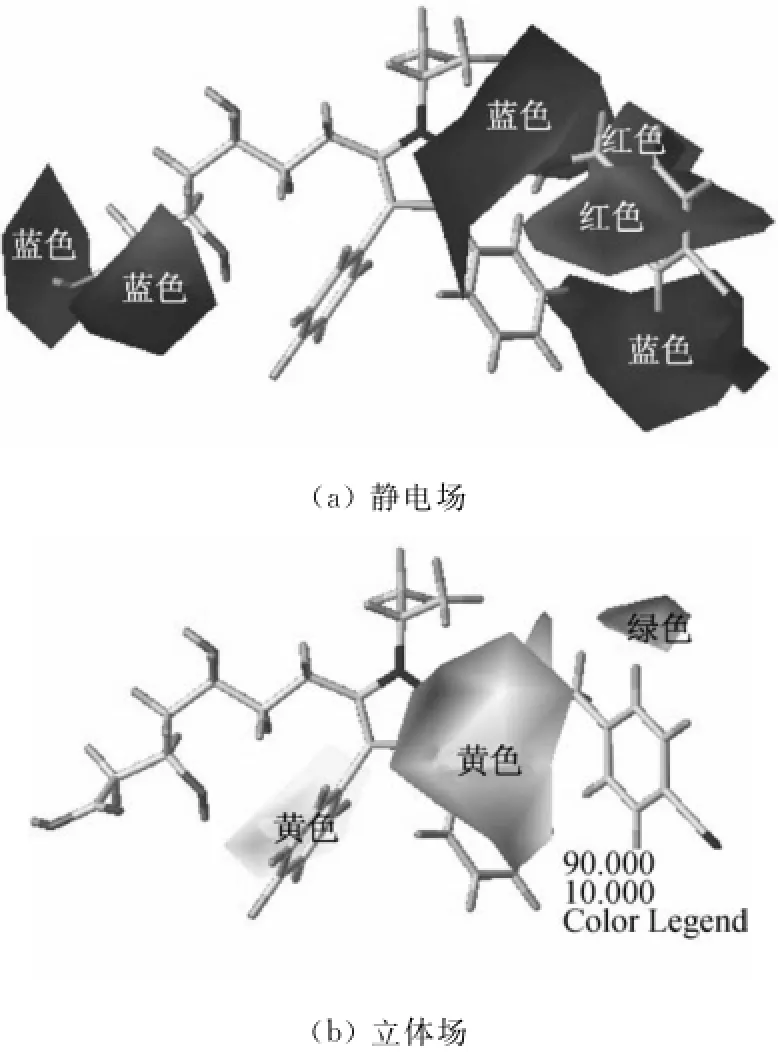
图5 Co MFA模型的等值图Fig.5 The contour maps of CoMFA model
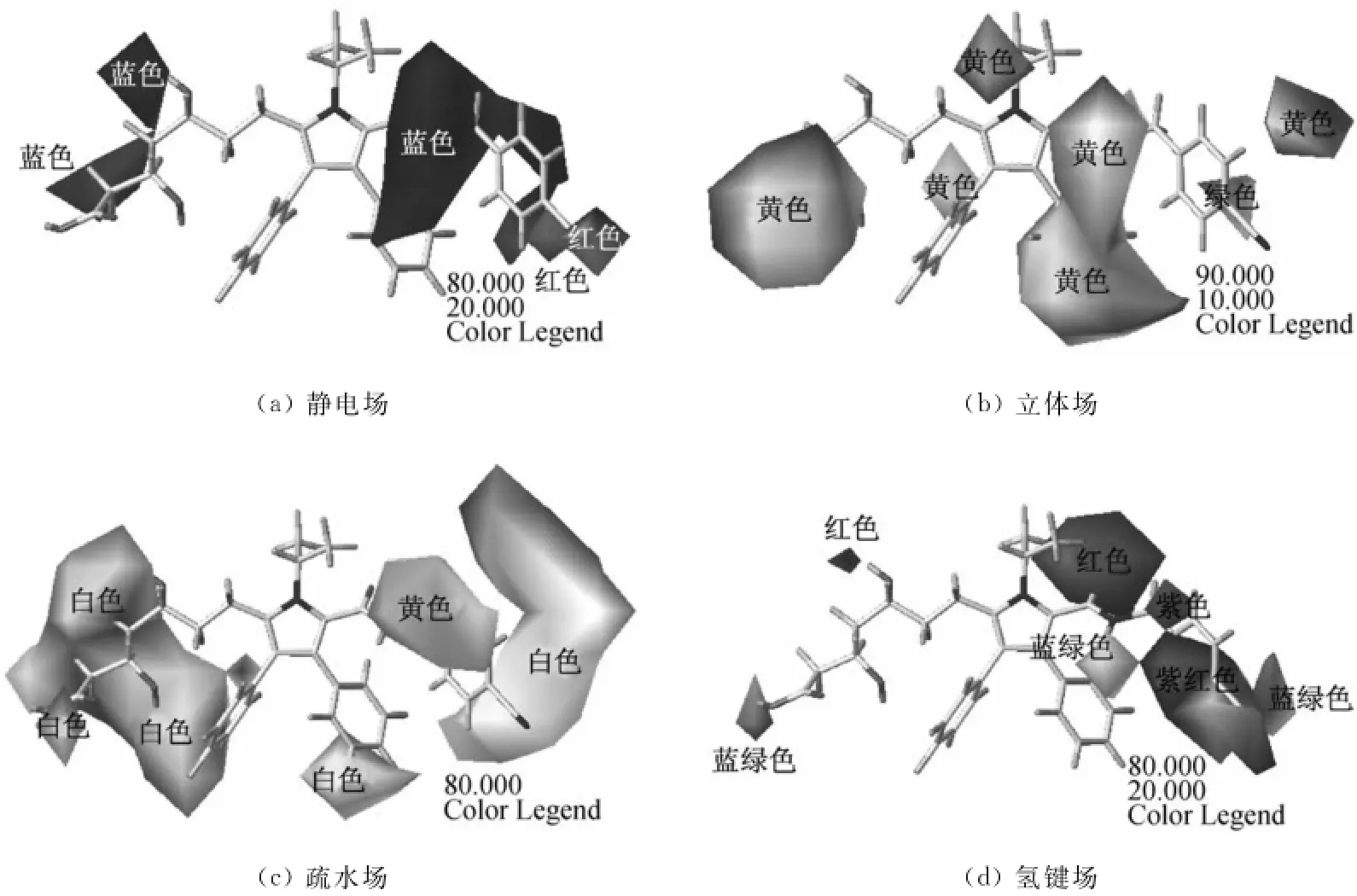
图6 CoMSIA模型的等值图Fig.6 The contour maps of Co MSIA model
2.2 分子对接
由SYBYL软件中的柔性对接计算结果如图7所示,抑制剂分子与HMGR蛋白的相互作用非常好,模板分子(22号分子)与HMGR蛋白中的7个氨基酸残基形成了氢键作用.这7个氨基酸都位于结合口袋内部,Lys735、Ser684和Arg590与脂肪链的羧酸基中羰基和羟基上的氧原子形成氢键,Lys691、Lys692、Asp690和Glu559分别与脂肪链上3,5位的羟基形成了氢键作用.抑制剂分子结构中的A环5位上芳香环取代基,B、C环分别与HMGR蛋白中疏水性氨基酸残基形成疏水共轭作用,使得抑制剂可以更好地结合HMGR,延长药物作用时间.综上可知,可以合理修饰抑制剂分子结构,得到更高活性、低毒性的抑制剂分子.
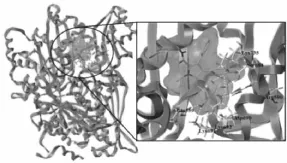
图7 分子对接Fig.7 Molecular docking
2.3 模型的验证
提取分子对接后与对接前的构象做一个叠合比对来探讨模型的可靠性,本文中选取活性最好的分子22作为模板分子进行叠合比对,叠合图如图8所示,A是对接后的构象,B是对接前的构象.由图可见,药效团(phamacophore)部分叠合良好,而其他部分因位于结合口袋袋口,与受体相关蛋白形成重要的疏水作用,故这些部分的结构相差较大,但是对活性不起主导作用.综上所述,分子对接前后的构象在药效团部分构象叠合较好,分子对接与3DQSAR相互支持,故分子对接前的构象可以作为3D-QSAR模型建立的结构,而所建的模型是可靠的,可以作为设计高活性HMGR抑制剂分子的有用模型.
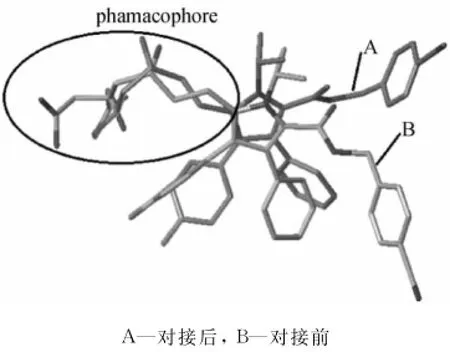
图8 对接前后的分子叠合图Fig.8 The alignment figure of anteroposterior molecular dock
3 结 语
3D-QSAR模型和分子对接可从不同角度认识新型Atorvastatin类HMGR抑制剂,根据文献可知,脂肪链是其药效部位,脂肪链上的羧酸和羟基可以与相关氨基酸的侧链残基形成许多氢键.因氟原子是电负性最大的元素,且在体内代谢缓慢,可延长药物的半衰期,故若在脂肪链的合适位置引入氟原子或含氟基团,可相对地改善活性.对于35个抑制剂分子建立的最优3D-QSAR模型,根据得到的不同力场等值图,可以合理修饰原有的分子结构,从不同位置修饰结构,可提高活性、减少用药量,相对地降低药物毒性.
[1] Grundy S M.Cholesterol and coronary heart disease:anew era[J].J Am Med Assoc,1986,256(20):2849-2858.
[2] Thelle D S.Hypercholesterolaemia[J].Drug Invest,1990,2(2):1-8.
[3] 杨亚莉.降血脂新药-西立伐他汀从市场撤出[J].中国药事,2001,15(5):62.
[4] Cheng L P,Huang X Y,Wang Z,et al.Combined 3D-QSAR,molecular docking,and molecular dynamics study on potent cyclohexene-based influenza neuraminidase inhibitors[J].Monatsh Chem,2014,145(7):1213-1225.
[5] Wang Z,Cheng L P,Kai Z P,et al.Molecular modeling studies of atorvastatin analogues as H MGR inhibitors using 3D-QSAR,molecular docking and molecular dynamics simulations[J].Bioorg Med Chem Lett,2014,24(16):3869-3876.[6] Pfefferkorn J A,Choi C,Song Y T,et al.Design and synthesis of novel,conformationally restricted HMG-CoA reductase inhibitors[J].Bioorg Med Chem Lett,2007,17(16):4531-4537.
[7] Pfefferkorn J A,Song Y T,Sun K L,et al.Design and synthesis of hepatoselective,pyrrole-based H MG-Co A reductase inhibitors[J].Bioorg Med Chem Lett,2007,17(16):4538-4544.
[8] Bratton L D,Auerbach B,Choi C,et al.Discovery of pyrrole-based hepatoselective ligands as potent inhibitors of H MG-Co A reductase[J].Bioorg Med Chem,2007,15(16):5576-5589.
[9] Clark M,Cramer R D,Van Opdenbosch D,et al. Validation of the general purpose tripos 5.2 force field[J].J Comput Chem,1989,10(8):982-1012.
[10] Gasteiger J,Marsili M.Iterative partial equalization of orbital electronegativity:a rapid access to atomic charges[J].Tetrahedron,1980,36(22):3219-3228.
[11] Abdulhameed M D M,Hamza A,Liu J J,et al. Combined 3D-QSAR modeling and molecular docking study on indolinonederivativesasinhibitors of 3-phosphoinositide-dependent protein kinase-1[J].J Chem Inf Model,2008,48(9):1760-1772.
[12] Park W K C,Kennedy R M,Larsen S D,et al. Hepatoselectivity of statins:design and synthesis of 4-sulfamoyl pyrroles as H MG-Co A reductase inhibitors[J].Bioorg Med Chem Lett,2008,18(3):1151-1156.
(编辑 吕丹)
3D-QSAR and Molecular Docking of Novel Atorvastatin Derivatives as HMGR lnhibitors
WANG Zhi, CAI Mingfeng, YE Liangchun, LIU Zhuoyu, CHENG Liping
(School of Chemical and Environmental Engineering,Shanghai Institute of Technology,Shanghai 201418,China)
Hydroxymethylglutaric acyl coenzyme A reductase(HMGR)is an important catalytic ratelimiting enzyme for the synthesis of endogenous cholesterol.The synthesis of endogenous cholesterol can be reduced by inhibiting the activity of H MGR.H MGR inhibitors(statins)are one of the best drugs available for treating the cardiovascular diseases(CVD).Combining three-dimensional quantitative structure-activity relationship(3D-QSAR)and molecular docking analysis and with the application of the computer aided drug design(CADD),the relationship between the synthesized HMGR inhibitor structures and inhibitory activity was studied.On the basis of the optimal 3D-QSAR model and the integrated molecular docking,a new type of HMGR inhibitors molecule with higher activity was designed.
statin;three-dimensional quantitative structure-activity relationship(3D-QSAR);molecular docking;hydroxymethylglutaric acyl coenzyme A reductase(HMGR)
R 914.5
A
1671-7333(2015)04-0327-06
10.3969/j.issn.1671-7333.2015.04.003
2014-11-05
上海市自然科学基金资助项目(15ZR1440400);上海市大学生科技创新创业资助项目(PE2014044,PE2014069);上海应用技术学院本科毕业设计重点资助项目(33110T145016)
王 志(1987-),男,硕士生,主要研究方向为制药工程.E-mail:yzhiwang1987@163.com
程利平(1974-)女,副教授,博士,主要研究方向为计算机辅助新药设计及合成,药物定量构效关系.
E-mail:chengliping@sit.edu.cn
