Huntington’s disease:a tutorial review
2015-11-02JeanMarcBurgunder
Jean-Marc Burgunder
Huntington’s disease:a tutorial review
Jean-Marc Burgunder
Huntington’s disease is a rare autosomal-dominant disorder,affecting people in middle age with motor,cognitive and psychiatric symptoms. The disease is due to a triplet repeat elongation in the Huntingtin gene,which leads to neuronal malfunction and degeneration through a number of different molecular pathways.Molecular genetic testing,which is performed after careful neurogenetic counselling,confirms diagnosis.The treatment is symptomatic and needs to be tailored to the need of the patients and involve his relatives.
In this tutorial,major aspects of the clinical presentation,pathophysiology and treatment of this disorder are briefly reviewed.
1 Introduction
Huntington’s disease is an autosomal-dominant disorder with a complex and variable phenotype comprising motor,cognitive and psychiatric features.It is fully penetrant and leads to death after a relentless progressive course of the symptomatology over 15-20 years.In the last couple of years,a tremendous increase on our knowledge of the disorder has taken place,mainly through the efforts of research groups including the Huntington Study Group(HSG)and the European Huntington’s disease Network(EHDN). During the same time,and specifically after the discovery of the gene involved in this disease allowing its investigation in cellular and animal models,a similar increase in our understanding of the pathophysiology involved has been witnessed.In this tutorial,basic knowledge on clinical presentation,pathophysiology and treatment of Huntington’s disease is presented.
2 Phenotype
There are two phases in the assessment of the phenotype,the first is at time of first investigation,when the examination should search for the major symptoms and signs to allow the diagnosis of chorea with cognitive and psychiatric symptoms,as well as include a full neurological and general examination for the precise differential diagnostic analysis.The second phase is during the course of the disorder,when examination is more concerned with recognition and understanding of the present complains expressed by patient and family in order to provide symptomatic relief.Both may be complemented by assessment in the frame of observational studies with specific protocols,like the EHDN Registry Study,or Enroll-HD.
The hallmark feature of the disease is chorea(Ta-ble 1),which means dance in the Greek,and the disorder had been accordingly named Huntington’s chorea for a while,but other symptoms may affect function even more than chorea,and chorea is not the only motor disorder experience by these patients.Therefore the wording Huntington’s disease is now preferred.
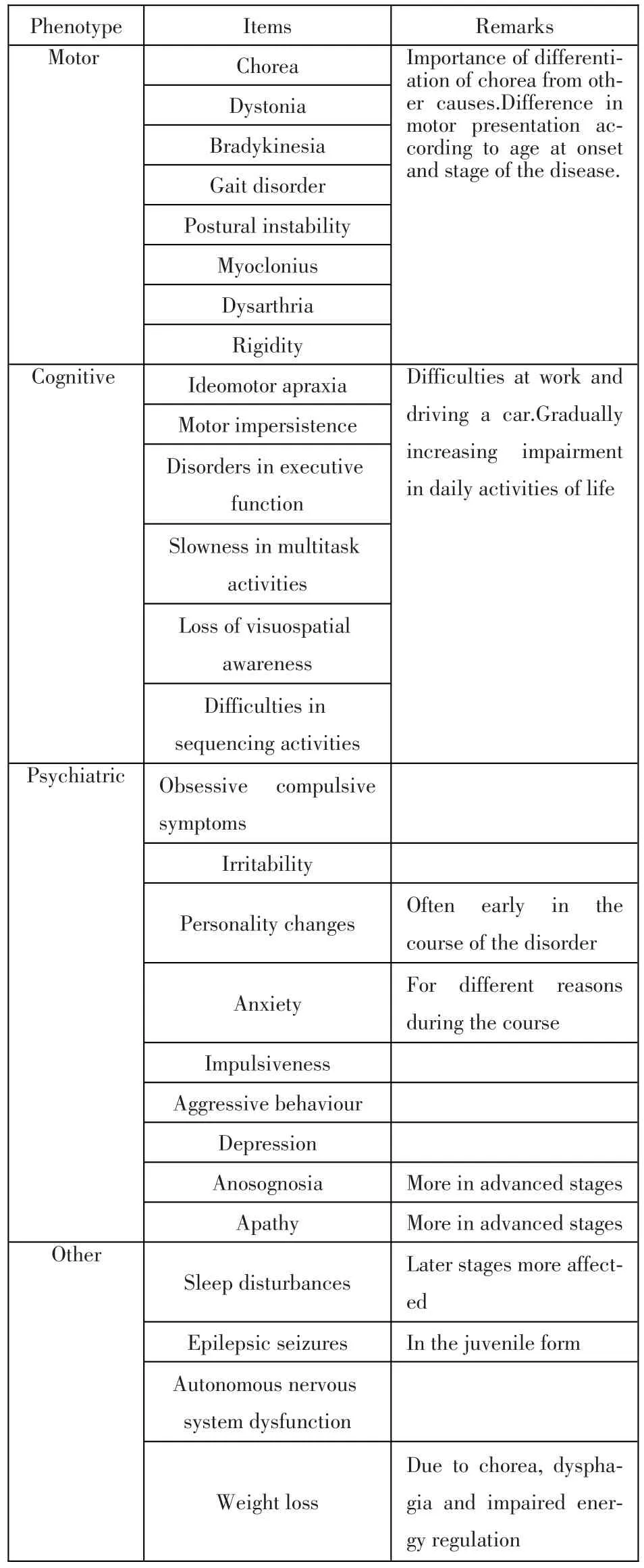
Table1 The phenotype of Huntington’s disease
Chorea in itself resembles levo-dopa induced dyskinesia,indeed a study using multisensor magnetic tracker system with full body recording,revealed similarities in a number of parameters,and only subtle differences were found for speed of movement[4].It is important to assess in a systematic fashion other potential causes of chorea,especially in patients with atypical presentation or absent family history(Table 2).
Cognitive symptoms(Table 1)may predate the appearance of chorea for some years.In a cohort of people at risk for HD,40%had mild cognitive impairment,with a tendency to increase closer to disease manifestation[5].These early changes have profound functional consequences,and early function decline is predicted by motor,cognitive and depressive symptoms[6].The type of cognitive impairment is different from other neurodegenerative disorders,like Parkinson’s or Alzheimer’s disease.In early stages of the disorder,impairment is found in single domains rather than a generalised fashion[5].Processing speed and episodic memory are affected early in the course of the disorder,executive function impairment is also predominant in early stages[7].Patients may also have defects in emotion recognition,typically in recognising disgust.This may severely impair their social integration.There is a predominant disorder of executive functions[8].Symptom severity is often not recognised by HD patients,who sometime even demonstrate anosognosia.As the disease progresses dementia frequently worsens,but even in severe cases some aspects of cognition may remain recognizable.
The psychiatric symptoms(Table 1)in HD may be very variable and represent a complex of different neuropsychological mechanisms related directly and as a reaction to the disease process impairing brainfunction.Anxiety about the disease in the family and the perception of the risk of the disease,and episodes of depression may occur long before onset of motor symptoms.This may be followed by subtle changes in personality,with aggressive behaviours and disinhibition.Suicide is a frequent complication in HD.

Table2 The major other disorders in the differential diagnosis of chorea
Sleep is disturbed in people affected with HD[9],latency is prolonged,efficiency decreased,nocturnal awakening increased and deep,slow-wave sleep decreased.Some of the features described are specific to HD as compared to other neurodegenerative disorders[10].These changes are correlated with both the severity of the other clinical presentation and of brain morphological changes,however,in early cases they seem not to be recognized by the patients[11].
The patients typically live through a number of stages in their affection by HD.It starts with the recognition of the family disorder,which may occur quite early in adolescence,goes through the question of knowing their own genetic status,to the fear of now getting symptomatic and to the different stages of impairment.The course of the disease is a constant progression of the symptoms with death typically 15 to 20 years after diagnosis[12].
Extending from the clinical examination run for the diagnostic process,there is a need to perform precise assessment and documentation during the course of the disease.This is a problem-oriented approach,which should inform about the need of the patient in order to offer him specific therapies according to his present needs.The United HD rating scale(UHDRS)captures motor,functional,behaviour and cognitive symptoms[13],and has the to be complemented by precise instructions,including video with the original publication[13]and also on line(www.euro-hd.net,for EHDN registered members).
对不同设计水平下的组合进行模拟试验,以均值7.66为临界值,获得大于临界值的方案49个,各变量取值的频率分布见表7。
3 Diagnosis
A careful history and examination will typicallyallow the classification of movement disorder and its cause[14].Information should be carefully collected on different aspects,including family history(Table 3). Family history over several generations with a particular attention about consanguinity should be taken precisely.Some pitfalls must be kept in mind,including non-paternity and secrecy about disease involvement for psychosocial or other reasons.The examination will have the purpose to confirm the presence and characterisation of the movement disorder and help to disclose additional features including sensory,pyramidal and neuromuscular involvement.

Table3 Investigations in the diagnostic p rocedures
In the presence of an autosomal dominant inheritance of a subtle onset and gradually progressive syndrome including chorea,cognitive and behavioural changes,which can present in a variable fashion even among members of the same family,the diagnosis of Huntington’s disease is the most probable one.In such a case there is no need of any addition investigation and a genetic test may be contemplated,especially in the case this has not yet been done in another member of the family[15].This has to be performed with great care during a genetic counselling workout ideally including the family,since such a result has important implications for the patients and his relatives,and thoughtful consideration of the information provided and the way it is received is mandatory.In the case of a negative testing,which is a rare occurrence with this typical presentation,at least in Western populations[16],additional information may guide further diagnosis(Table 2).Many of the sporadic disorders with chorea may be diagnosed by precise history and examination.Sometimes relatively simple laboratory tests may help further(Table 3).Imaging studies and neurophysiological investigations may be needed in selected patients(Table 3)
4 Epidem iology
The prevalence of the disorder is quite variable and values in Western populations are given between 4 and 8 individual per 100 000 of population[17].In a recent comparative meta-analysis,the prevalence was 0.40 in China/Japan,and 5.7 in Europe/North America and Oceania[18].In a review of cases published in the Chinese literature[19]only small number of cases had been previously reported.However,centres collaborating in the Chinese Huntington’s Disease Network are now seeing increasing number of cases.It may be,that the prevalence figures will have to be revised in the future.
5 Course of the disorder
Most often,people affected by HD have a profound experience of the disorder from their family.Already as children,they may start to be recognized,that there is this pecular disorder in their families. Gradually,relatives of patients with the disorders,specifically children of affected parents,start to askthemselves about their own risk.Later,they may contemplate genetic testing in order to know for themselves.In the case they turn out to carry the gene mutation,there is a long time of uncertainty,whether subtle symptoms they may experience are already manifestations of the disorder.Later,they start to experience impairment at work and in their daily life,subtle changes in mood and behaviour may be recognised by their relatives.Over the years,functional abilities are gradually lost until the need of increasingly intensive care arises,which poses a great burden on the family.In the more advanced stages of the disorder,bodily functions,like feeding,sphincter control,also get gradually lost.At the same time communication is getting more and more difficult,due to increasing dysarthria.Death is often due to pneumonia,however during earlier phases of the disorder,suicide is not rare.
6 Genetics
Huntington’s disease is an autosomal dominant disorder and the gene has been found after a complex search years after the locus had been recognised[20]. This gene encodes huntingtin and contains a series of triplet CAG repeats in its first exon.The number of repeats is expanded in HD.Individuals with more than 36 CAG repeats will develop the disease,in a range of 36-39,the mutation is less penetrant.The number of triplet repeats is instable,and can change from one to the next generation.
Genetic testing must be performed with caution,especially in non-affected potential gene carrier[21]. High quality information must be provided in understandable words adapted to the counselee in order for him to be able to make his own decision.One major risk of genetic testing of healthy potential carrier is the occurrence of suicide and this is increased in people with a psychiatric history in the 5 years prior to the test and unemployed status[22,23].
7 Pathology
The major location of pathological change in the brains of people with HD is the caudate nucleus and putamen,followed by the cerebral cortex(predominantly the frontal parts),and globus pallidus,thalamus,hypothalamus[24,25].This leads to a severe generalised atrophy with loss of brain volume up to 30%as compared to age matched controls.Caudate involvement and decrease of brain volume can be measured already years before onset of obvious symptoms[26].
Several molecular mechanisms are involved to lead to neuronal dysfunction after mutation of the huntingtin,which is also expressed in the brain.The mutated protein may lead to gain and to loss of function. Accumulation of mutated huntingtin has been well established since some years[27],however,it is still debated,whether this is deleterious for neurons,or whether the process might have a protective effect against cytosolic mutated huntingtin toxicity[25].Huntingtin has a number of cellular functions,which may all be disturbed after mutation of the protein.This starts with impaired gene expression,which has been demonstrated for a number of genes related to neurotransmission in the striatum[28,29].A large number of molecular interactions are perturbated by the elongated glutamine repeat,leading to disturbance of intracellular protein trafficking[30].A major network of synergistic interactions with protein involved in mitochondrial stability and function may lead to energy crisis and neurodegeneration[31].Mutation in huntingtin disturbs the gene regulation of several proteins involved in mitochondrial function,and the mutated may react with mitochondrial membrane proteins,impairing respiratory chain function,anchoring of mitochondria to microtubules,mitochondrial fusion and fission dynamics and increasing calcium transport.They may also impair autophagy,increase apoptosis,modify neurotrophic supply,intracellular solute biosynthesis and intercellularsignalling.

Table4 M ajor treatm ent m odalities in HD
8 Treatment
There are several types of therapies in HD,which can be classified among symptomatic and causal therapies.Symptomatic therapies are targeted towards directly modifying the present objective signs and subjective complains of the patient.They can be further separated according to the different major syndrome complexes in motor,psychiatric and cognitive therapies and studies available classified as HD specific and non-HD specific.Causal therapies are included in direct gene therapies and other,molecular-based indirect therapies.The first are aimed at directly targeting the mutated gene or its transcript as the single cause of the disorder.The second are aimed at correcting the pathways in the complex sequence of molecular and neuronal plastic changes leading to the expression of the disorder.Causal therapies are now not available,but intensive research is now being pursued in order to modify the disease process by acting on the different molecular processes described above[32].
In the meantime,it is important to realise,that much can be done for the care of patients with HD and to support their families and carers.In doing so,it is important to tailor the symptomatic treatment to the need of the patient.The best provision of care will be in a multidisciplinary setting.Before onset and in the first stage of the disorder,the interdisciplinary team will include neurologist,psychiatrist and geneticist,later physiotherapist,speech and occupational therapists,nursing and other medical specialists according to the multiple and increasing needs of gradually dependent patients.Palliative care and end of life stage management gets increasing attention[33].Only few clinical therapeutic trials have assessed symptomatic treatment in HD[34],and the evidence available to provide suggestions for the symptomatic treatment on HD is poor[35].Therefore we must rely on expert opinion,sometimes more or less appropriately based on studies on chorea or psychiatric symptoms in patients suffering from other disorders.The major aim remains to improve quality of life.Even for the most obvious aspects of the phenotype,movement disorder,only spare data on treatment are available[36-37],and we have recently performed a survey on the use of drugs to treat chorea in different continents[38].Patient’s need,including stigma,physical injury,gait instability,work interference,and disturbed sleep are indications to start antichoreic drugs,with different choicesof the drugs available(Table 4).Antipsychotic drug is preferred when comorbid psychotic or aggressive behaviours are present.Tetrabenazine is only one drug approved by the USA food and drug administration for the treatment of chorea in HD[39].The major mechanism of tetrabenazine action is the inhibition of a vesicular monoamine transporter(VMAT).Tetrabenazine,at adjusted doses up to 100 mg per day,decreases chorea significantly[40],and discontinuation of the drug is followed by worsening of chorea[41].Side effects must be considered,they include bradykinesia with tremor,and,for tetrabenazine,depression.Antipsychotic drug may includ,for example,olanzapine(2.5~10 mg),risperidone(0.5~2 mg),or tiapridal(50~200 mg).Doses need to be adapted to the response,and,sometimes,it may be useful to try several antipsychotic drugs to find the most appropriate on in a selected patient.In the presence of disabling myoclonus,valproate may be used[42].Movement disorders in late disease stages may respond to baclofen or benzodiazepines.Chemodenervation using botulinum toxin injection in hyperactive muscles may sometimes be useful for focal spasms,including bruxism or focally predominant spastic overactivity.Physiotherapy is also important,and a recent study has provided evidence,that focused training of posture and gait is beneficial for patients with HD[43].Provisional guidelines with suggestions for patients in each stage of the disorder have recently been presented[44].
There are only few studies on pharmacological treatment of cognitive impairment in HD,and none has shown any benefit.Assessment is important,in order to counsel the patients and family about occupation adaptation if still working and also about the development of coping strategies.Cognitive therapy may be useful in order to help the patient and his environment to structure activities and manage available resources.Environmental strategies may also be of value in advanced cases.
Evidence to base treatment of psychiatric symptoms specifically in HD is very poor,and most guidelines rely on personal expertise and on suggestions extrapolated from studies in disorders with a mostly psychiatric phenotype.Based on expert views surveys about obsessive compulsive behaviours[45],and irritability[46],a step-wise procedure can be suggested,with a serotonin reuptake inhibitor as the first choice,combined with behavioural therapy,at least in the patients with only mild cognitive impairment.In the presence of additional depression,anxiety or obsessive-compulsive behaviours a serotonin reuptake inhibitor is suggested as first line.For both groups,behavioural management strategies are advisable,this may include protection from stress factors in advanced cases in the need of institutionalised chronic care.After dose optimisation the next step would be combination therapy.Similar approaches may be used for the treatment of depression.
Dysphagia occurs in later stage of the disease and is due to the involuntary orofacial movement disorder,the decrease in motor control,the propensity to eat rapidly and side effects of drugs,including xerostoma due to anticholinergic effects.There is no controlled study to guide choices in dysphagia,but established methods include providing swallowing tips(which should be started before cognitive impairment precludes learning),to prepare food in appropriate ways and provide them in a quite and supervised environment.Gastric feeding needs to be discussed early,in order to understand patient’s choices and the danger of choking and aspiration pneumonia as potential causes of complications.Weight loss is frequent in HD and is due to dysphagia,choreatic movement but also to a modulation of metabolism.It is important to appropriately increase energy intake in a way suitable for the patient.
There are a number of socio-medical problems faced by the patients and their relatives,which needs to be addressed in appropriate ways by trained professionals.
9 Conclusion:towards the future
HD is a severe disease,but it is wrong to think,that nothing can be done against it.A number of symptomatic treatments are now available and comprehensive care programmes show their benefit for the patients and their families.Research and care programmes are now in place with networks of clinicians,researchers,lay organisations.The European Huntingon’s disease network is an example of such an effort,and more than 10 000 persons affected with HD and controls have been included so far in the Registry cohort study,the major EHDN study.A global study,Enrol-HD,sponsored by CHDI,has been started some months ago.In China researchers,clinicians and persons affected with HD have founded the Chinese HD Network and started several collaborative research projects.
[1]Walker FO.Huntington's Disease[J].Semin Neurol,2007,27(9557):143-150.
[2]Nicolas G,Dodier D,Alice G,et al.Juvenile Huntington disease in an 18-month-old boy revealed by global developmental delay and reduced cerebellar volume[J].Am J Med Genet A,2011,155(4):815-818.
[3]Lefaucheur R,Maréchal LG,Wallon D,et al.Chorea in an 83-year-old woman:don't forget Huntington's disease[J].J Am Geriatr Soc,2012,60(5):983-984.
[4]Mann RK,Edwards R,Zhou J,et al.Comparing movement patterns associated with Huntington's chorea and Parkinson's dyskinesia[J].Exp Brain Res,2012,218(4):639-654.
[5]Duff K,Beglinger LJ,Theriault D,et al.Cognitive deficits in Huntington's disease on the Repeatable Battery for the Assessment of Neuropsychological Status[J].J Clin Exp Neuropsychol,2010,32(3):231-238.
[6]Beglinger LJ,O'Rourke JJ,Wang C,et al.Earliest functional declines in Huntington disease[J].Psychiatry Res,2010,178(2): 414-418.
[7]Wahlin TBR,Lundin A,Dear K.Early cognitive deficits in Swedish gene carriers of Huntington's disease[J].Neuropsychology,2007,21(1):31-44.
[8]Paulsen JS.Cognitive impairment in huntington disease:diagnosis and treatment[J].Curr Neurol Neurosci Rep,2011,11(5): 474-483.
[9]Aziz NA,Anguelova GV,Marinus J,et al.Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington's disease[J].Parkinsonism Relat Disord,2010,16(5):345-350.
[10]Petit D,Gagnon JF,Fantini ML,et al.Sleep and quantitative EEG in neurodegenerative disorders[J].J Psychosom Res,2004,56(5):487-496.
[11]Goodman AOG,Barker RA.How vital is sleep in Huntington's disease?[J].J Neurol,2010,257(6):882-897.
[12]Roos RA,Hermans J,Vlis VVD,et al.Duration of illness in Huntington's disease is not related to age at onset[J].J Neurol Neurosurg Psychiatry,1993,56(1):98-100.
[13]Kremer HPH,Hungtington SGX.Unified Huntington's Disease Rating Scale:reliability and consistency.Huntington Study Group[J].Mov Disord,1996,11(2):136-142.
[14]Wild EJ,Tabrizi SJ.The differential diagnosis of chorea[J]. Pract Neurol,2007,7(6):360-373.
[15]Harbo HF,Finsterer J,Baets J,et al.EFNS guidelines on the molecular diagnosis of neurogenetic disorders:general issues,Huntington's disease,Parkinson's disease and dystonias[J].Eur J Neurol,2009,16(7):777-785.
[16]Wild EJ,Tabrizi SJ.Huntington's disease phenocopy syndromes[J].Curr Opin Neurol,2007,20(6):681-687.
[17]Harper PS.The epidemiology of Huntington's disease[J].Hum Genet,1992,89(4):365-376.
[18]Pringsheim T,Katie W,Lundy D,et al.The incidence and prevalence of Huntington's disease:a systematic review and meta-analysis[J].Mov Disord,2012,27(9):1083-1091.
[19]Zheng Z,Burgunder JM,Shang H,et al.Huntington's like conditions in China,A review of published Chinese cases[J].PLoS Curr,2012,4:RRN1302.
[20]Macdonald ME,Ambrose CM,Duyao MP,et al.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes[J].Cell,1993,72(6): 971-983.
[21]Tibben A.Predictive testing for Huntington's disease[J].Brain Res Bull,2007,72(2-3):165-171.
[22]Almqvist EW,Bloch M,Brinkman R,et al.A worldwide assess-ment of the frequency of suicide,suicide attempts,or psychiatric hospitalization after predictive testing for Huntington disease[J].Am J Hum Genet,1999,64(5):1293-1304.
[23]Almqvist EW,Brinkman RR,Wiggins S,et al.Psychological consequences and predictors of adverse events in the first 5 years after predictive testing for Huntington's disease[J].Clin Genet,2003,64(4):300-309.
[24]Vonsattel JP,DiFiglia M.Huntington disease[J].J Neuropathol Exp Neurol,1998,57(5):369-384.
[25]Waldvogel HJ,Kim EH,DC Thu,et al.New Perspectives on the Neuropathology in Huntington's Disease in the Human Brain and its Relation to Symptom Variation[J].Journal or Huntington's Disease,2012,1:1-11.
[26]Tabrizi SJ,Reilmann R,Roos RA,et al.Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study:analysis of 24 month observational data[J].Lancet Neurol,2012,11(1):42-53.
[27]DiFiglia M,Sapp E,Chase KO,et al.Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain[J].Science,1997,277(5334):1990-1993.
[28]Luthi-Carter R,Apostol BL,Dunah AW.et al.Complex alteration of NMDA receptors in transgenic Huntington's disease mouse brain:analysis of mRNA and protein expression,plasma membrane association,interacting proteins,and phosphorylation[J].Neurobiol Dis,2003,14(3):624-636.
[29]Luthi-Carter R,Strand A,Peters NL,et al.,Decreased expression of striatal signaling genes in a mouse model of Huntington's disease[J].Hum Mol Genet,2000,9(9):1259-1271.
[30]Ratovitski T,Nakamura M,D'Ambola J,et al.N-terminal proteolysis of full-length mutant huntingtin in an inducible PC12 cell model of Huntington's disease[J].Cell Cycle,2007,6(23): 2970-2981.
[31]Costa V,Scorrano L.Shaping the role of mitochondria in the pathogenesis of Huntington's disease[J].Embo J,2012,31(8): 1853-1864.
[32]Burgunder JM.Translational research in Huntington's disease: opening up for disease modifying treatment?[J].Translational Neurodeg,in press.
[33]Marks S,Hung S,Rosielle DA.Palliative care for patients with Huntington's disease#201[J].J Palliat Med,2011,14(5): 655-656.
[34]Nance MA.Therapy in Huntington's disease:where are we?[J]. Curr Neurol Neurosci Rep,2012,12(4):359-366.
[35]Mestre T,Coelho MM,Costa J,et al.Therapeutic interventions for symptomatic treatment in Huntington's disease[J].Cochrane Database Syst Rev,2009(3):CD006456.
[36]Bonelli RM,Wenning GK.Pharmacological management of Huntington's disease:an evidence-based review[J].Curr Pharm Des,2006,12(21):2701-2720.
[37]Bonelli RM,Hofmann P.A systematic review of the treatment studies in Huntington's disease since 1990[J].Expert Opin Pharmacother,2007,8(2):141-153.
[38]Burgunder JM,Guttman M,Perlman S,et al.An International Survey-based Algorithm for the Pharmacologic Treatment of Chorea inHuntington's Disease[J].PLoSCurr,2011,3: RRN1260.
[39]Poon LH,Kang GA,Lee AJ.Role of tetrabenazine for Huntington's disease-associated chorea[J].Ann Pharmacother,2010,44(6):1080-1089.
[40]HS Group.Tetrabenazine as antichorea therapy in Huntington disease:a randomized controlled trial[J].Neurology,2006,66(3):366-372.
[41]Frank S,Ondo W,Fahn S,et al.A study of chorea after tetrabenazine withdrawal in patients with Huntington disease[J].Clin Neuropharmacol,2008,31(3):127-133.
[42]Saft C,Lauter T,Kraus PH,et al.Dose-dependent improvement of myoclonic hyperkinesia due to Valproic acid in eight Huntington's Disease patients:a case series[J].BMC Neurol,2006,6(1):11.
[43]Bohlen S.Physical therapy in Huntington's disease-toward objective assessments?[J].Eur J Neurol,2012.
[44]Quinn L,Busse M.Physiotherapy clinical guidelines for Huntington's disease[J].Neurodegen Dis Manage,2012,2:21-31.
[45]Anderson K,Craufurd D,Edmondson MC,et al.An International Survey-based Algorithm for the Pharmacologic Treatment of Obsessive-Compulsive Behaviors in Huntington's Disease[J]. PLoS Curr,2011,3:RRN1261.
[46]Groves M,Van DE,Anderson K,et al.An International Survey-based Algorithm for the Pharmacologic Treatment of Irritability in Huntington's Disease[J].PLoS Curr,2011,3:RRN1259.
10.3969/j.issn.1002-0152.2015.10.002
Swiss Huntington’s Disease Centre,Department of Neurology,University of Bern
