替米沙坦片含量的专属性试验
2015-10-25孔凯丽
孔凯丽 成 芳
(江苏天士力帝益药业有限公司,江苏 淮安 223003)
替米沙坦片含量的专属性试验
孔凯丽成 芳
(江苏天士力帝益药业有限公司,江苏 淮安 223003)
目的 验证替米沙坦片含量测定方法的专属性。方法 建立了HPLC法(用十八烷基硅烷键合硅胶为填充剂;以0.005 mol/L氢氧化钠甲醇溶液为溶剂;以2.0 g/L磷酸二氢铵溶液(用磷酸调节pH值至3.0)-甲醇(30∶70)为流动相;检测波长为298 nm;柱温40 ℃;流速1.0 mL/min;理论板数按替米沙坦峰计算不低于4000。测定替米沙坦片及其降解产物含量。考察高温、高湿、光照、酸、碱破坏对替米沙坦片的影响,确定方法专属性情况。结果 替米沙坦片在高温、高湿、光照、酸、碱破坏条件下稳定,在氧化破坏条件下部分降解。结论 所建立的HPLC方法能够较好的考察替米沙坦片在高温、高湿、光照、酸、碱破坏下的降解情况,方法专属性良好。
替米沙坦片;降解;专属性
替米沙坦片是一种口服起效的,对其他受体无亲和力,用于原发性高血压的治疗。替米沙坦片的含量测定方法,国家食品药品监督管理局批准的标准和美国药典收载的标准均采用HPLC法,但美国药典样品处理时直接取20片,经溶剂溶解后,过滤,稀释,进样;而国家局批准的标准采用研磨后,称取适量,溶解、过滤后进样。
替米沙坦片在制备过程中,首先在碱性溶液中替米沙坦和葡甲胺形成替米沙坦葡甲胺钠盐,葡甲胺极易溶解于水,并具有引湿性,导致在研磨过程易吸附于器壁上,并导致研磨不均。替米沙坦片现行的质量标准中的含量测定检测方法不完善,不能准确的体现含量测定结果,准确度、精密度较差。为了准确的体现测定结果,我们参考美国药典中替米沙坦片含量测定检测方法进行研究验证。结果表明,美国药典收载的替米沙坦片含量测定结果的准确度和精密度较好。我们按照《已上市化学药品变更研究的技术指导原则》要求,参照美国药典收载的替米沙坦片含量测定方法,进行相关的研究验证工作。
1 仪器与试剂
clarus 600气相色谱仪;安捷伦GC-MS 6890N/5973气质联用仪。岛津高效液相色谱仪(SCL-10AVp);色谱柱:Diamonsil,C18(2),50 mm×4.6 mm,5µm。HHS电热恒温水浴锅(江苏省医疗器械厂);CM-3型澄明度检测仪(天津药典标准仪器厂);BT125D(十万分之一)电子天平(赛多利斯科学仪器有限公司);替米沙坦片(江苏天士力帝益药业有限公司自制,批号:20140312);替米沙坦片对照品(99.9%,供含量测定用,LOT:100629-201202,中国药品生物制品检定所);替米沙坦有关物质A对照品:(供含量测定用,批号:F01326,USP ROCKVILU.MD);磷酸二氢铵(分析纯,批号:F20110530,国药集团化学试剂有限公司);氢氧化钠(分析纯,批号:12050810742,南京化学试剂有限公司);甲醇(HPLC,批号:13020444,TEDTA);磷酸(HPLC,批号:PA-3102-1000,ACS)。
2 实验方法
照高效液相色谱法(中国药典2010年版二部附录VD)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;以0.005 mol/L氢氧化钠甲醇溶液为溶剂;以2.0 g/L磷酸二氢铵溶液(用磷酸调节pH值至3.0)-甲醇(30∶70)为流动相[1];检测波长为298 nm;柱温40 ℃;流速1.0 mL/min;理论板数按替米沙坦峰计算不低于4000。
测定法:取本品20片,置200 mL量瓶中,加溶剂适量,振摇至分散,超声10 min,放冷至室温,加溶剂至刻度,摇匀,滤过,精密量取续滤液适量,加流动相稀释制成每1 mL中约含0.12 mg的溶液,精密量取10 µL,注入液相色谱仪,记录色谱图。另取替米沙坦对照品12 mg,置100 mL量瓶中,加流动相适量超声使溶解[2],用流动相稀释至刻度,摇匀,同法测定。按外标法以峰面积计算,即得。
3 降解试验
3.1空白溶剂:取溶剂、流动相、空白辅料各10 µL进样,记录色谱图,空白溶剂不干扰替米沙坦含量测定(图1)。
3.2未破坏样品:取替米沙坦片(20140312批)20片,置200 mL量瓶中,加溶剂适量超声10 min,放冷至室温,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,无杂质峰干扰替米沙坦含量测定(图2)。
3.3高温破坏:取替米沙坦片(20140312批)20片,置200 mL容量瓶中,水浴加热30 min,冷却至室温,加溶剂适量,超声10 min,用溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,替米沙坦在高温条件下与未破坏样品比较基本无变化(如未破坏样品图谱)。
3.4氧化空白: 取过氧化氢溶液(30%)5.0 mL置200 mL容量瓶中,沸水浴10 min,冷却至室温,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,无杂质峰(图3)。
3.5氧化破坏:取替米沙坦片(20140312批)20片,置200 mL量瓶中,加过氧化氢溶液(30%)1.0 mL,摇匀,置沸水浴中10 min后,取出,放却,加溶剂适量超声10 min,放冷至室温,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,替米沙坦在氧化条件下与未破坏样品比较基本无变化[3](如未破坏样品图谱)。
3.6酸碱空白:吸取1 mol/L盐酸溶液和1 mol/L氢氧化钠溶液各5.0 mL置200 mL量瓶中,混匀,置沸水浴中10 min后,放冷,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,酸碱空白溶液几乎没有干扰(图4)。
3.7酸破坏:取替米沙坦片(20140312批)20片,置200 mL量瓶中,加入5.0 mL 1mol/L盐酸溶液,再置沸水浴中10 min后,放冷,加入5.0 mL 1mol/L氢氧化钠溶液中和,加溶剂适量超声至溶解,用溶剂定容至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,替米沙坦在酸性条件下与未破坏样品比较基本无变化(如未破坏样品图谱)。
3.8碱破坏:取替米沙坦片(20140312批)20片,置200 mL量瓶中,加入5.0 mL 1mol/L氢氧化钠溶液,再置沸水浴中10 min后,放冷,加入5.0 mL 1mol/L盐酸溶液中和,加溶剂适量超声10 min,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,替米沙坦在碱性条件下与未破坏样品比较基本无变化(如未破坏样品图谱)。
3.9光照破坏:取替米沙坦片(20140312批)20片,置200 mL量瓶中,于4500LUX下照射24 h,取出,加溶剂适量超声10 min,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,替米沙坦在光照条件下未发生变化(如未破坏样品图谱)。
3.10高湿破坏:取替米沙坦片(20140312批)20片,置表面皿中,于高湿条件下(92.5%RH)24 h,用溶剂转移至200 mL量瓶中,加溶剂适量超声10 min,加溶剂至刻度,摇匀,滤过,精密量取续滤液3 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,精密量取10 µL,注入液相色谱仪,记录色谱图,替米沙坦在高湿条件下与未破坏样品比较基本无变化(如未破坏样品图谱)。
4 结 论
替米沙坦片在高温、氧化、酸、碱、高湿、光照条件下稳定,替米沙坦片含量检查方法专属性良好[4]。

图1

图2

图3
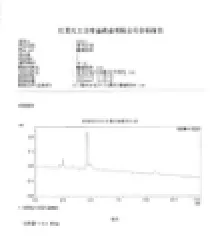
图4
[1]秦宗会,夏红梅,谢兵,等.甲酚红分光光度法测定替米沙坦的含量[J].理化检验(化学分册),2012,48(1):90-92.
[2]谢兵,何以恒,谭蓉,等.固绿FCF分光光度法测替米沙坦的含量[J].分析试验室,2011,30(2):74-77.
[3]甘湘庆,秦宗会.偶氮胂Ⅰ分光光度法测定替米沙坦的含量[J].光谱实验室,2010,27(6):2191-2196.
[4]谭蓉,秦宗会,马新会,等.酚红分光光度法测定替米沙坦的含量[J].理化检验(化学分册),2010,46(4):390-392.
R917
B
1671-8194(2015)20-0048-02
