利用CRISPR/Cas9技术对人多能干细胞进行高效基因组编辑
2015-10-22刘改改李爽韦余达张永贤丁秋蓉
刘改改,李爽,韦余达,张永贤,丁秋蓉
中国科学院上海生命科学研究院营养科学研究所,上海 200031
利用CRISPR/Cas9技术对人多能干细胞进行高效基因组编辑
刘改改,李爽,韦余达,张永贤,丁秋蓉
中国科学院上海生命科学研究院营养科学研究所,上海 200031
CRISPR/Cas9技术提供了一个全新的基因组编辑体系。本文利用CRISPR/Cas9平台,在人胚胎干细胞株中对选取的一段特定基因组区域进行了多种基因组编辑:通过在基因编码框中引入移码突变进行基因敲除;通过单链DNA提供外源模板经由同源重组定点敲入FLAG序列;通过同时靶向多个位点诱导基因组大片段删除。研究结果表明CRISPR/Cas9可以对多能干细胞进行高效基因编辑,获得的突变干细胞株有助于对基因和基因组区域的功能进行分析和干细胞疾病模型的建立。
CRISPR/Cas9基因编辑;人多能干细胞;基因敲除;DNA序列定点插入;基因组大片段删除
网络出版时间: 2015-9-1611:06:06
URL: http://www.cnki.net/kcms/detail/11.1913.R.20150916.1106.004.html的基因突变和疾病发生具有强烈相关性[1]。深入解析这些遗传突变,并建立携带特定遗传突变的疾病模型用于针对性的药物筛选,将极大提高对疾病致病机理的认识和加速疾病防治方案的开发。
人类多能干细胞(Human pluripotent stem cells,hPSCs)和基因组编辑技术结合所建立的细胞模型,为疾病研究提供了一个独特的实验平台。利用这个平台体系,研究人员可以研究特定基因突变甚至染色体结构变异对人类多种细胞类型和组织器官功能的影响及其详细的分子机制,并可建立携带不同遗传突变的“个性化”疾病模型用于大规模药物筛选。该模型体系的建立得益于基因组编辑技术,尤其是CRISPR/Cas9(Clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins9,CRISPR/ Cas9)技术的飞速发展。目前常用的CRISPR/ Cas9平台是从化脓链球菌(Streptococcus pyogenes)的Ⅱ型CRISPR系统优化而来,由SpCas9核酸酶和gRNA构成[2~5]。其中SpCas9核酸酶识别基因组中的PAM (Protospacer adjacent motif)序列,gRNA中长约20 bp的引导序列决定靶向特异性。当基因组上位于PAM 5′端的前体间隔序列(Protospacer)与gRNA中的引导序列互补时,Cas9核酸酶启动对DNA双链进行切割,形成DNA双链断裂缺口(Double-strand breaks,DSBs)。DSBs能激活细胞内非同源末端连接(Non-homologous end joining, NHEJ)和同源重组(Homology directed repair, HDR)两种DNA修复机制。NHEJ修复过程可以引入碱基的随机插入或者缺失(Insertion or deletion);而在外源模板存在的情况下,细胞也可以通过HDR的方式对基因组进行精确修复。利用CRISPR/Cas9技术,本实验室建立了在人多能干细胞中进行基因敲除或者敲入的基因组编辑体系[6,7]。本研究以位于人2号染色体上的LINC00116基因组区域为例,利用CRISPR/Cas9技术对人多能干细胞中的该基因组区域进行了基因敲除、FLAG短肽序列定点插入和基因组大片段删除,获得的多个突变干细胞株为下一步对该基因组区域进行功能分析提供了特有的细胞平台。
1 材料和方法
1.1CRISPR/Cas9载体构建
通过Online软件(http://www.genome-engineering.org)针对特定基因组区域进行CRISPR靶向序列设计和脱靶位点预测,或者通过对靶向位点附近序列进行观察,寻找符合要求的序列,进一步通过序列比对确认特异性后获得靶向序列。将靶向序列(20 bp)克隆入pgRNA_cloning vector (Addgene,http://www. addgene.org/)获得gRNA质粒。pCas9-GFP质粒来自Addgene。文中用到的靶向序列(下划线为PAM)分别是:ORF靶向:5′-GTGCTAGTAGCCTTCGCTTCTGG-3′;FLAG定点敲入:5′-GGCGGAAGGCGCAGAGTCTCAGG -3′;大片段删除:5′-GTAACACATGCCCAGAGGCAGGG -3′和5′-GCTAAGCCTCCAGTACAATGTGG -3′。FLAG-ssODN(Singlestranded oligodeoxynucleotide)序列为(下划线为FLAG编码序列):5′-GCAAGGTGGCCATGAGGGGGACAGAATGGGGCGGAAGGCGCAGAGTCT CACTTGTCATCGTCATCCTTGTAATC GGCCAGG TCCAGCTTCTTCTGCGTCGCCGCCAGCTTGTCC TGCAGCCTC-3′,购自美国IDT(Integrated DNA Technoliges)公司。
1.2gRNA靶向活性鉴定
为鉴定gRNA的靶向活性,Cas9质粒分别和不同的gRNA质粒共转入HEK293T细胞。48 h后对细胞进行基因组DNA抽提,并对靶向位点附近的基因组序列进行PCR扩增(产物长度为300~500 bp)。PCR产物经过纯化后利用Surveyor Mutation Delection试剂盒(Transgenomic,USA)进行活性分析。
1.3hPSCs培养和电穿孔
人胚胎干细胞株HUES9[8]在mTeSR1培养基(STEMCELL Technologies,Canada)中培养。培养皿预先用Geltrex(Life Technologies,USA)处理,每24 h更换新鲜mTeSR1培养基,细胞密度达80%时用Accutase(STEMCELL Technologies,Canada)消化传代。
细胞在电穿孔前用10 μmol/L ROCK 抑制剂(Santa Cruz Biotechnologies,USA)预处理3~4 h,随后用Accutase消化为单细胞悬液。对细胞计数后将1×107个细胞重悬至800 μL预冷的PBS中,加入25 μg pCas9-GFP质粒和25 μg gRNA表达质粒或者15 μg pCas9-GFP质粒、15 μg gRNA表达质粒和30 μg FLAG-ssODN后混合。将约800 μL细胞与DNA混合液转移至0.4 cm电穿孔杯(Bio-Rad, USA)中,冰上孵育5 min,在电穿孔仪(Bio-Rad, USA)中用250 V和500 μF的条件进行电穿孔。电穿孔后加入适量培液进行细胞重悬,再度离心去除死细胞后种下。
1.4细胞分选和单克隆分离
电穿孔48 h后,荧光显微镜下观察绿色荧光蛋白(GFP)以确定CRISPR/Cas9质粒成功表达。细胞用Accutase消化为单细胞悬液后离心重悬于PBS中,通过流式分选出绿色荧光表达细胞,以每块10 cm培养皿15~30 K个细胞的密度种下。细胞恢复生长3~4 d后镜下可见单克隆,10 d左右单克隆被挑选分离至96孔板中。当96孔板中细胞密度达到80%左右时,进行细胞消化传代,获得两块同样的96孔板细胞,一块用于继续培养,另一块贴壁恢复生长一段时间后用于基因组DNA抽提和基因型鉴定。
1.5基因型鉴定
用于基因型鉴定的96孔板在细胞密度长至80%~ 90%时,PBS洗去培养基后每孔加入50 μL细胞裂解液(10 mmol/L Tris pH 7.5,10 mmol/L EDTA,10 mmol/L NaCl,0.5% Sarcosyl,40 μg/mL蛋白酶K)在56~60℃孵育过夜裂解细胞。随后每孔加入100 μL预冷的95%乙醇和75 mmol/L NaCl在室温或-20℃孵育1 h进行离心沉淀DNA。75%乙醇漂洗2遍后室温晾干,每孔用30 μL的ddH2O或者TE缓冲液加0.1 mg/mL RNase A进行DNA溶解。
溶解获得的基因组DNA用于下一步PCR鉴定基因型。设计PCR引物对靶向位点附近的基因组DNA进行扩增,扩增片段长度为150~200 bp。PCR产物首先通过2.5%的琼脂糖胶电泳鉴定,携带NHEJ引入的碱基插入或缺失或者通过HDR定点插入FLAG序列的PCR产物在胶上会表现出位移。潜在阳性单克隆的PCR产物通过Sanger测序进一步确认序列或者克隆到T载体中测序确认序列。
2 结果与分析
2.1干细胞基因编辑体系建立
本课题组前期利用CRISPR/Cas9技术构建了一个对人多能干细胞株进行基因组编辑的平台体系(图1)[6,7,9]。该体系包含两个质粒,一个是由CAG(CMV early enhancer/chicken beta actin promoter)启动的SpCas9表达质粒,并以2A肽段连接的方式同时表达GFP荧光蛋白,作为下游筛选标记;另一个质粒包含U6启动的gRNA元件,可以通过简单的酶切连接方式获得具有特定靶向序列的gRNA质粒。实验流程主要包括电穿孔、流式分选、单克隆形成、单克隆基因型鉴定和扩增等步骤。同批实验的多个突变克隆和野生型克隆可用于后期研究。
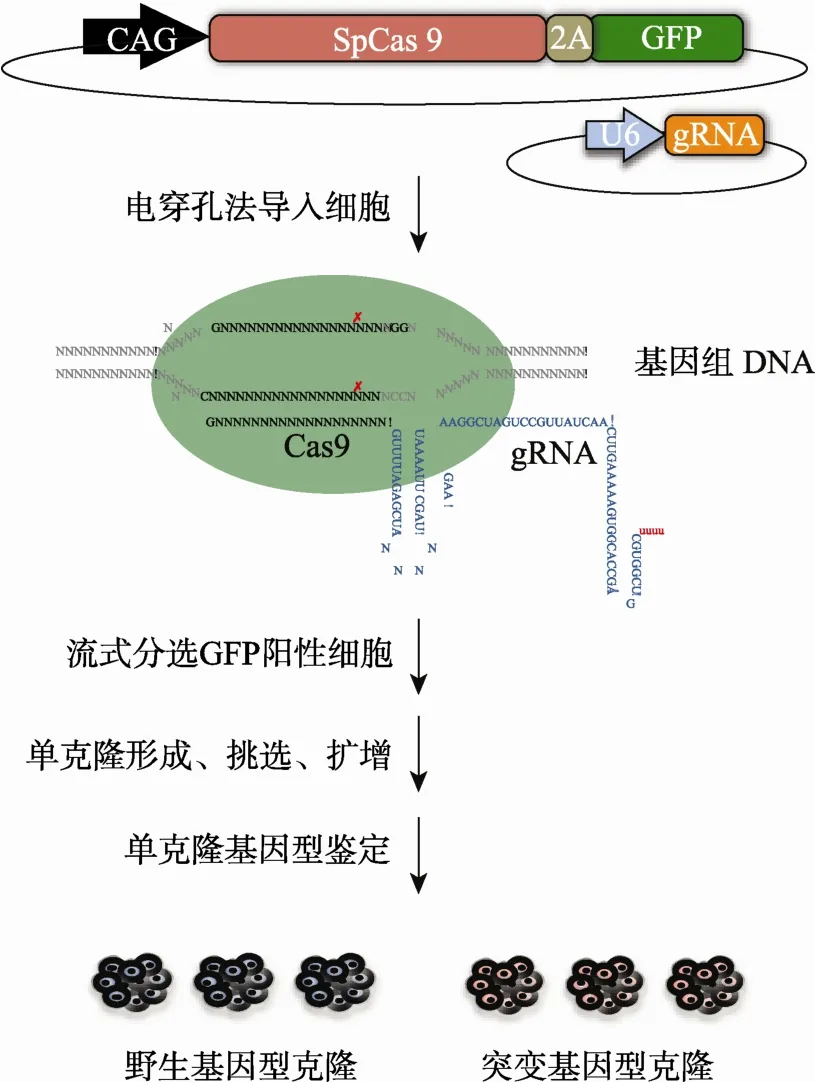
图1 利用CRISPR/Cas9靶向人多能干细胞流程示意图
2.2基因敲除
近期一项针对小鼠中长链非编码RNA(Long intergenic noncoding RNA, lincRNA)的大规模筛选表明,若干非编码RNA和脂肪分化紧密相关,其中一条名称为lnc-RAP-5[10]。LINC0016为lnc-RAP-5人种属中相应的同源基因,它位于人2号染色体上,总长约15 kb。通过对这段基因组区域的基因编码框和物种保守性分析,发现在LINC00116基因第7个外显子中存在一个物种保守性很高的基因编码框(Open reading frame, ORF),提示在前期研究中被预测为非编码的LINC00116基因组区域有可能编码蛋白。为探究这种可能性,确认该ORF是否编码蛋白,以及其在脂肪分化中可能的作用,本文首先利用CRISPR/Cas9靶向该编码区,通过引入移码突变获得基因敲除干细胞株(图2A)。靶向后共获得162个单克隆。进一步对获得的单克隆进行靶向基因组区域的PCR扩增和测序,成功鉴定出93个突变克隆(突变率约57%),其中双等位基因具有相同序列突变的纯合子克隆有14个(纯合率约8.6%),双等位基因带有不同突变的复合型杂合子克隆有9个(复合杂合子率约5.6%),剩余单等位基因突变杂合子克隆有70个(单突变杂合子率约43%)。对突变克隆的序列分析显示突变一般发生在位于PAM 5′端的3个碱基附近,碱基插入或缺失的数目均小于20 bp,绝大部分在10 bp以内,最常见的为1 bp碱基插入和5 bp碱基缺失(图2B)。
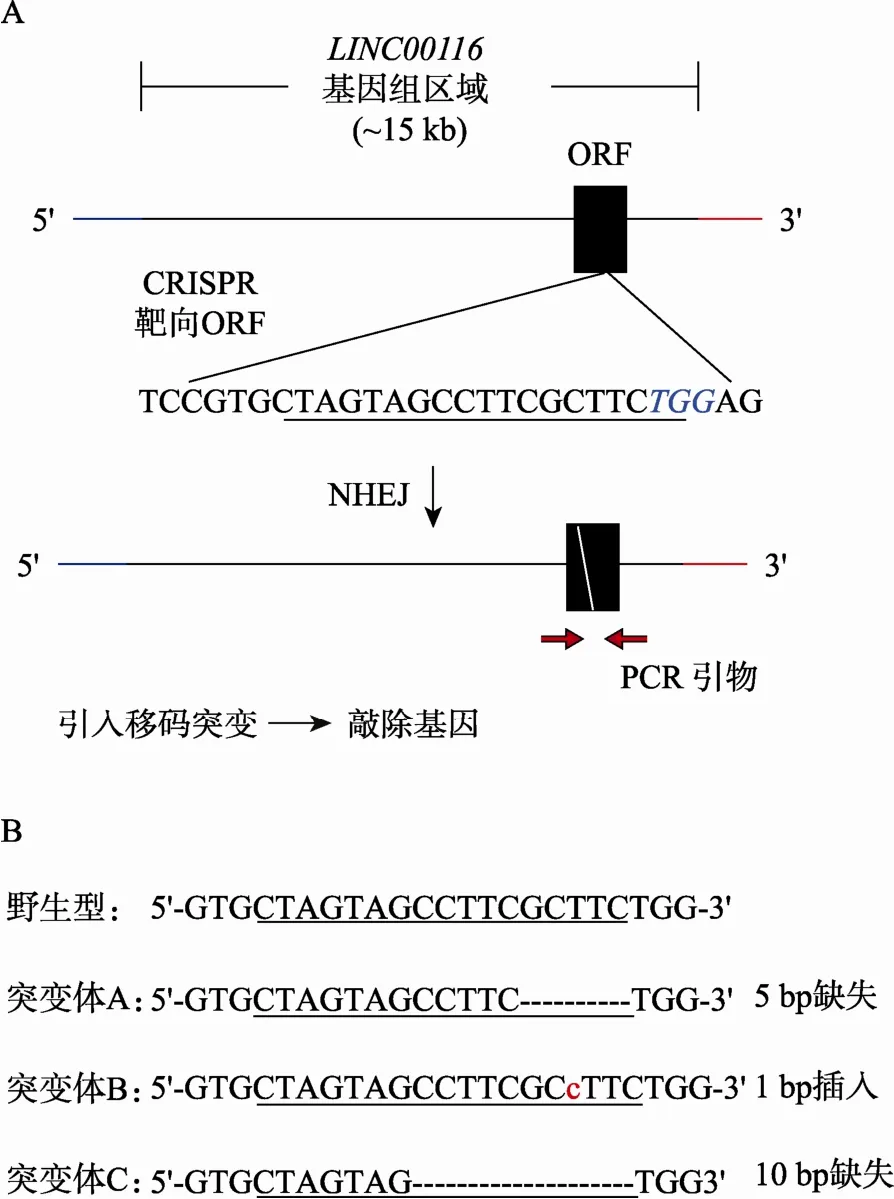
图2 利用CRISPR/Cas9敲除基因
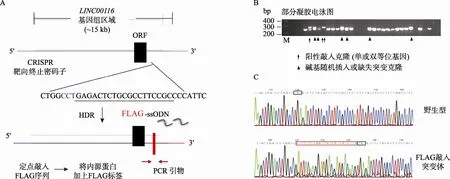
图3 利用CRISPR/Cas9定点敲入FLAG序列
2.3FL AG短肽序列的定点敲入
由于缺乏合适的内源蛋白抗体,为确定该基因编码框是否编码蛋白,本文在终止密码子前定点敲入编码FLAG小肽的DNA序列,用FLAG小肽标记内源蛋白,因而后期可以通过FLAG抗体对内源蛋白的表达和功能进行检测。gRNA序列靶向终止密码子,在HEK293T细胞中的活性检测中显示较高的靶向活性(附图1)。单链核苷酸FLAG- ssODN携带FLAG编码序列以及靶向序列两边长为50 bp的同源臂序列作为外源模板。Cas9-GFP、gRNA和FLAG-ssODN同时导入多能干细胞进行基因编辑(图3A)。靶向后共获得450个单克隆。通过PCR对靶向位点附近的基因组序列进行扩增后发现成功插入FLAG序列(24 bp)的克隆在琼脂糖电泳中相比野生型克隆显示明显的位移(图3B),也同时存在若干经由NHEJ修复过程引入碱基插入或缺失突变而产生的突变克隆(图3B)。随后通过对潜在阳性克隆进行测序进一步确认FLAG序列的正确插入(图3C)。最终鉴定结果显示1共获得5个阳性克隆,其中一个克隆的两条等位基因中均有正确FLAG序列插入,定点插入效率约1.1%。
2.4基因组大片段删除
为同时研究LINC00116的生物学功能,以及LINC00116和ORF编码蛋白之间的生物学联系,本文将全部LINC00116基因组区域(约15 kb)进行大片段删除后观察对细胞功能的影响。两条gRNAs序列分别靶向LINC00116基因组区域两边,在HEK293T细胞的活性检测中均显示出较高的靶向活性(附图1)。将两条gRNA同时导入细胞后,基因组区域两边产生DSBs,有部分细胞因而发生大片段缺失(图4A),靶向后一共获得190个克隆。通过PCR进行阳性克隆筛选,首先在靶向位点的两边设计PCR引物(第1对引物),发生大片段缺失的阳性克隆能扩增出PCR产物,野生型克隆由于中间序列很长而不会有PCR产物生成(图4B,上方电泳图);随后通过针对大片段中间序列设计引物(第1对引物)对获得的阳性克隆中大片段缺失是发生在1条等位基因还是同时发生在两条等位基因中进行分析(图4B,下方电泳图)。结果显示在11个单克隆中发生了大片段缺失(阳性率约5.6%),其中一个单克隆的两条等位基因中均发生了片段缺失(图4B)。PCR产物的测序结果进一步确认了大片段缺失(图4C)。
3 讨 论
本研究以位于人2号染色体上的LINC00116基因组区域为例,通过CRISPR/Cas9基因组编辑技术成功在人胚胎干细胞株HUES9中进行了多种基因编辑:通过在基因编码框中引入移码突变敲除基因;通过单链核苷酸提供外源同源模板经由同源重组定点敲入FLAG序列;通过同时导入两条gRNAs诱导基因组大片段缺失。各种突变效率比较显示通过NHEJ过程引入碱基插入或缺失产生移码突变的效率最高,在本研究中效率为57%,其中在两个等位基因中同时发生碱基插入或缺失的突变效率为14%。利用CRISPR对多能干细胞中多个基因组区域进行靶向的研究结果显示,经由NHEJ引入碱基插入或缺失突变的效率均大于50%[6],提示利用CRISPR技术可以进行高效的基因敲除,甚至多个基因的同时敲除。
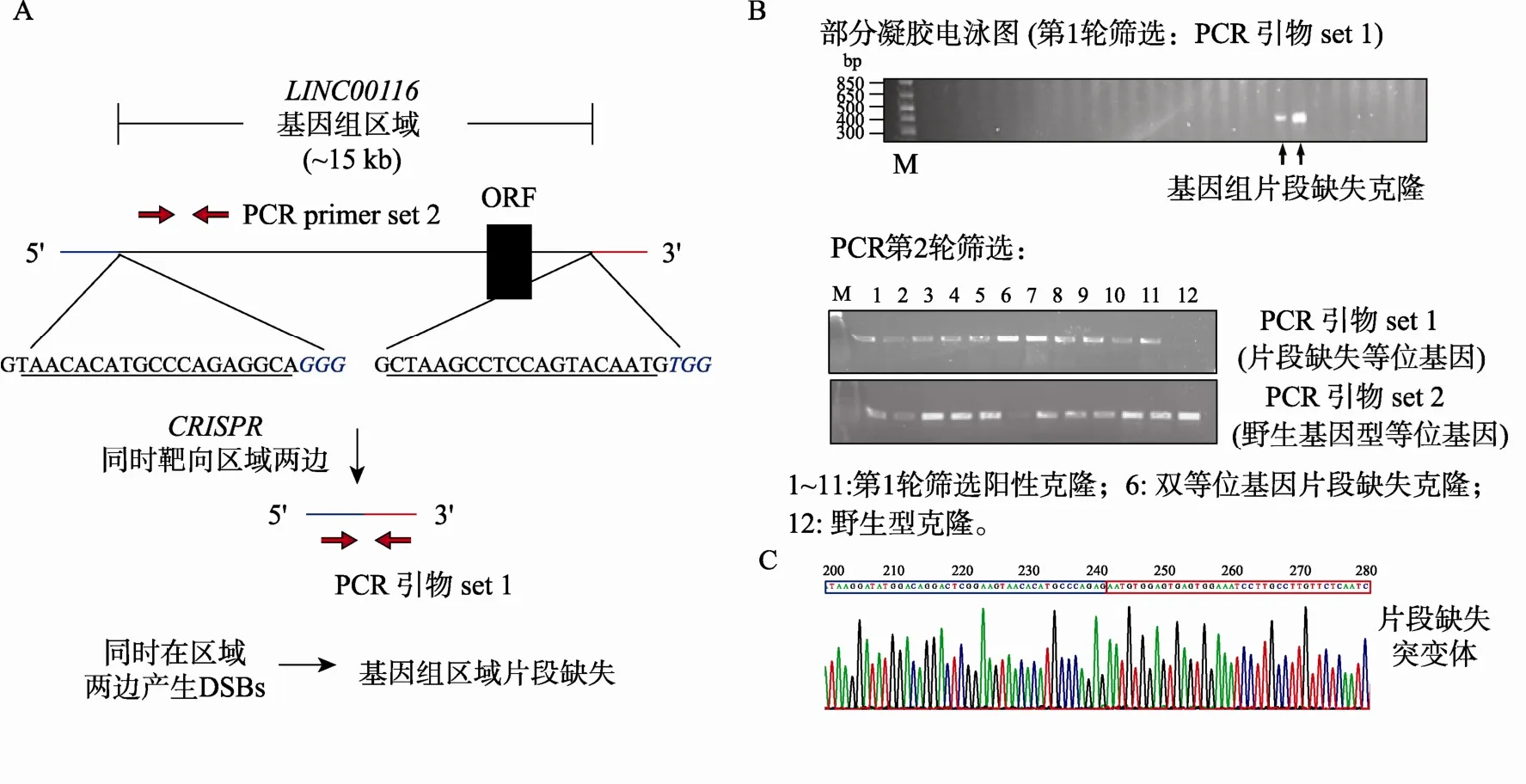
图4 利用CRISPR/Cas9进行基因组大片段删除。
在由同源重组敲入特定点突变或者外源序列方面,本研究通过单链核苷酸模板定点敲入FLAG小肽序列的效率偏低,仅为1.1%。后期或可尝试从以下几个方面提高重组效率:(1)增加单链核苷酸模板中同源臂的长度。本文中单链核苷酸模板两边同源臂长度为50 bp,但受单链核苷酸体外合成长度的限制(目前≤200 bp),同源臂长度一般小于100 bp;(2)构建质粒载体作为外源模板。利用质粒载体可以大幅度增加同源臂长度,并且可以引入抗性基因表达框经由药物筛选富集成功重组的细胞。本实验室前期通过两边携带500 bp长度同源臂和嘌呤霉素(Puromycin)筛选基因的质粒载体成功在多个基因组位点定点插入GFP报告基因,阳性克隆效率>60%(未发表数据);(3)提高gRNA的靶点活性。筛选更高活性的gRNA靶向序列可能有助于提高重组效率;(4)抑制NHEJ过程。由于细胞在DNA修复过程中NHEJ和HDR是两个相互拮抗的过程,抑制NHEJ过程可以相应提高HDR效率。多个研究组已经成功通过体外筛选找到抑制NHEJ修复过程的小分子化合物[11~13]。在基因靶向同时通过此类小分子化合物处理细胞,可以达到增强HDR过程而提高DNA序列敲入效率的目的。
本文利用同时导入两条gRNA对基因组区域进行了大片段靶向删除,效率约为5%。靶向删除基因组大片段的效率不仅和每条gRNA的基因编辑活性相关,同时也和片段长度相关。靶向删除片段长度的增加可能带来效率的降低[14]。此外,导入两条或者多条gRNAs,不仅可以引入基因组区域缺失,同时还可以引发其他多种染色体结构变异[15,16],包括染色体区域插入(Insertion)、重复(Duplication)、易位(Translocation)和倒位(Inversion)等,为模拟染色体结构变异导致的人类疾病提供了一个合适的平台。
基因靶向潜在的问题是脱靶效应。本研究组和其他研究组前期通过对靶向得到的干细胞单克隆株进行全基因组测序分析脱靶情况[17~19]。结果显示TALEN和CRISPR靶向后得到的干细胞克隆中脱靶率均很低,提示在保证靶向序列的高度特异性、控制基因编辑蛋白表达强度和时间的情况下,脱靶现象是可以避免的。值得注意的是,在全基因组测序中,研究人员发现很多单克隆(包括突变株和同批的野生型细胞株),相比编辑前的干细胞母株,产生若干新的点突变[9,17~19]。由于这些点突变附近基因组序列和靶向序列没有任何相似性,因此推测是在细胞培养和传代中随机发生,其中少数位于基因编码区而造成氨基酸突变。这种随机突变的获得提示基因靶向后获得的每个单克隆都可能存在靶向引入突变外的基因组序列上的差别。因而,在建立干细胞疾病模型对特定基因突变进行功能分析时,为避免这种随机突变对细胞表型的影响,实验设计需要对多个野生型和突变型克隆同时进行表型比对,在多个干细胞母株中进行疾病模型建立,并利用多种对照组设计(如在基因敲除克隆中通过引入外源质粒恢复基因表达)等进行综合分析。
CRISPR基因组编辑技术平台由于载体构建简单、靶向位点选择灵活、靶向效率稳定,目前已经被广泛用于各类细胞和模式动物的基因组编辑[20],它在干细胞平台中的应用潜力也远远超出了本文涉及的这几个方面。可以预见,基因组编辑技术和干细胞平台的结合将极大提高人们对生命科学的认识,推动精准医疗发展,加速细胞治疗和基因治疗等新一代医药方案的开发。
附录:附图1见电子版 www.chinagene.cn。
[1] The NHGRI GWAS Catalog. http://www.genome.gov/ gwastudies/.
[2] Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N,Hsu PD, Wu XB, Jiang WY, Marraffini LA, Zhang F. Multiplex genome engineering using CRIPSR/Cas systems. Science, 2013, 339(6121): 819-823.
[3] Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE,Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science, 2013, 339(6121): 823-826.
[4] Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. Elife,2013, 2: e00471.
[5] Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol, 2013, 31(3): 230-232.
[6] Ding QR, Regan SN, Xia YL, Oostrom LA, Cowan CA,Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell, 2013, 12(4): 393-394.
[7] Peters DT, Cowan CA, Musunuru K. Genome editing in human pluripotent stem cells. StemBook [Internet], 2013,Apr 29.
[8] Cowan CA, Klimanskaya I, McMahon J, Atienza J, Witmyer J, Zucker JP, Wang SP, Morton CC, McMahon AP,Powers D, Melton DA. Derivation of embryonic stem-cell lines from human blastocysts. N Engl J Med, 2004,350(13): 1353-1356.
[9] Ding QR, Lee YK, Schaefer EAK, Peters DT, Veres A,Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, Trevisan M, Gupta RM, Moisan A, Banks E,Friesen M, Schinzel RT, Xia F, Tang A, Xia YL, Figueroa E, Wann A, Ahfeldt T, Daheron L, Zhang F, Rubin LL,Peng LF, Chung RT, Musunuru K, Cowan CA. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell, 2013, 12(2): 238-251.
[10] Sun L, Goff LA, Trapnell C, Alexander R, Lo KA, Hacisuleyman E, Sauvageau M, Tazon-Vega B, Kelley DR,Hendrickson DG, Yuan BB, Kellis M, Lodish HF, Rinn JL. Long noncoding RNAs regulate adipogenesis. Proc Natl Acad Sci USA, 2013, 110(9): 3387-3392.
[11] Yu C, Liu YX, Ma TH, Liu K, Xu SH, Zhang Y, Liu HL,La Russa M, Xie M, Ding S, Qi LS. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell, 2015, 16(2): 142-147.
[12] Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K,Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol, 2015, 33(5): 543-548.
[13] Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol, 2015, 33(5): 538-542.
[14] Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, Maeda T, Paw BH, OrkinSH. Characterization of genomic deletion efficiency mediated by CRISPR/Cas9 in mammalian cells. J Biol Chem, 2014, 289(31): 21312-21324.
[15] Lee HJ, Kim E, Kim JS. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res,2010, 20(1): 81-89.
[16] Lee HJ, Kweon J, Kim E, Kim S, Kim JS. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res, 2012,22(3): 539-548.
[17] Veres A, Gosis BS, Ding QR, Collins R, Ragavendran A,Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell,2014, 15(1): 27-30.
[18] Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He CX,Wang Y, Brodsky RA, Zhang K, Cheng LZ, Ye ZH. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell, 2014, 15(1): 12-13.
[19] Suzuki K, Yu C, Qu J, Li M, Yao XT, Yuan TT, Goebl A,Tang SW, Ren RT, Aizawa E, Zhang F, Xu XL, Soligalla RD, Chen F, Kim J, Kim NY, Liao HK, Benner C, Esteban CR, Jin YB, Liu GH, Li YR, Izpisua Belmonte JC. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell, 2014, 15(1): 31-36.
[20] Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell, 2014,157(6): 1262-1278.
(责任编委: 张博)
Efficient genome editing in human pluripotent stem cells through CRISPR/Cas9
Gaigai Liu, Shuang Li, Yuda Wei, Yongxian Zhang, Qiurong Ding
Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Shanghai 200031, China
The RNA-guided CRISPR (clustered regularly interspaced short palindromic repeat)-associated Cas9 nuclease has offered a new platform for genome editing with high efficiency. Here, we report the use of CRISPR/Cas9 technology to target a specific genomic region in human pluripotent stem cells. We show that CRISPR/Cas9 can be used to disrupt a gene by introducing frameshift mutations to gene coding region; to knock in specific sequences (e.g. FLAG tag DNA sequence) to targeted genomic locus via homology directed repair; to induce large genomic deletion through dual-guide multiplex. Our results demonstrate the versatile application of CRISPR/Cas9 in stem cell genome editing, which can be widely utilized for functional studies of genes or genome loci in human pluripotent stem cells. Keywords: CRISPR/Cas9 genome editing; human pluripotent stem cells; gene disruption; targeted DNA insertion; largegenomic deletion
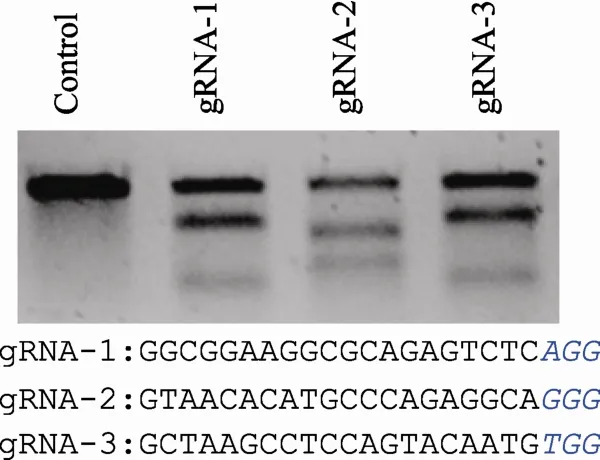
附图1 gRNA靶向活性鉴定。
2015-05-29;
2015-06-24
上海市浦江人才计划(编号:15PJ1409200)资助
刘改改,硕士,助理研究员,研究方向:干细胞治疗。E-mail: liugaigai@sibs.ac.cn
丁秋蓉,博士,研究员,研究方向:干细胞与转化医学。E-mail: qrding@sibs.ac.cn
10.16288/j.yczz.15-240
基因组测序技术的发展极大推动了人类对各类疾病遗传致病因素的认识。针对多种疾病进行的外显子测序(Exome sequencing)和全基因组关联研究(Genome-wide association study, GWAS)提示很多新
