商用SCR催化剂的钠中毒及再生
2015-10-12高凤雨唐晓龙易红宏赵顺征李东马玎张佟佟
高凤雨,唐晓龙,易红宏,赵顺征,李东,马玎,张佟佟
商用SCR催化剂的钠中毒及再生
高凤雨,唐晓龙,易红宏,赵顺征,李东,马玎,张佟佟
(北京科技大学土木与环境工程学院,北京,100083)
实验室模拟商用SCR催化剂(V2O5-WO3/TiO2)碱金属钠中毒,并通过水洗和0.5 mol/L H2SO4洗对失活催化剂进行再生。通过SEM,BET和XPS等方法对催化剂表面的理化性能进行分析,并探讨钒钛系催化剂的反应及失活机理。结果表明:较低负载量的Na2O会引发严重的催化剂中毒现象,通过水洗和酸洗再生可以使失活催化剂的表面活性得到不同程度的恢复,酸洗再生效果比水洗再生效果好;碱金属钠盐引起的催化剂物理中毒主要是盐颗粒的沉积和孔道的堵塞,但不是催化剂失活的主要原因,化学中毒主要是因为Na与催化剂表面的B酸性位上的V—OH发生反应,生成V—O—Na,改变催化剂表面金属氧化物的化学环境,阻碍NO和NH3向催化剂内部的扩散;催化剂的脱硝活性与其表面的化学吸附氧量和表面活性中心上(V4+)/(V5+)的比有一定的正相关性。
商用钒钛催化剂;钠中毒;再生;反应机理;失活机理
化石燃料的燃烧不可避免的产生大量的环境污染物质,其中,氮氧化物(NO:NO,NO2)是主要的大气污染物之一。排入大气中NO的90%产生于各种燃料燃烧过程,其中NO占95%左右。排放到大气中的NO会引起酸雨、光化学烟雾以及臭氧层的破坏,甚至对人体健康造成一定的损害作用[1−2]。目前,选择性催化还原(SCR)技术是应用最为广泛的脱硝技术,其中催化剂是SCR烟气脱硝技术的核心,广泛应用于SCR技术的商业催化剂是V2O5/TiO2基催化剂,并掺杂WO3或MoO3进行改性[3]。在SCR系统运行的过程中,催化剂会因为各种物理化学作用(如高温烧结、催化剂腐蚀/沾污/遮蔽、孔道堵塞、催化剂碱金属/碱土金属中毒、催化剂水和SO2中毒等)而失效[4−6]。其中,碱金属( 如K和Na等) 在较低含量的条件下就可以使催化剂严重失活[7−10]。目前,研究人员对SCR催化剂碱金属失活的研究多集中在K中毒方面,对Na中毒的报道较少,但其对催化剂的毒性作用是不可忽略的[11]。研究表明[12]:中毒后的SCR催化剂可以通过不同的方法来恢复其活性,从而实现催化剂的再生。本文作者以商用钒钛系SCR催化剂为研究对象,模拟Na2O对其毒性作用,探讨SCR催化剂的反应及其失活机理,并通过水洗和酸洗对失活催化剂进行再生,考察2种再生方法对失活催化剂的再生效果。
1 实验
1.1 新鲜催化剂预处理、中毒及再生
本研究所用催化剂为市售的蜂窝式整体催化剂,其主要成分为V2O5-WO3/TiO2。新鲜催化剂于研钵中研磨,取粒度为365~833 μm的催化剂颗粒于110 ℃烘干2 h备用。
以NaNO3为前驱体,采用超声波辅助浸渍法使新鲜催化剂颗粒表面负载一定量的Na2O,样品经1 h超声波浸渍后取出,于110 ℃干燥2 h,最后在空气气氛中于450 ℃焙烧3 h,即得失活催化剂样品。
分别采用水洗和H2SO4洗2种方法对失活催化剂样品进行再生。称取一定量的失活催化剂,分别置于适量的水和0.5 mol/L H2SO4溶液中浸渍1 h,取出,于110 ℃烘干,即得水洗和酸洗再生催化剂样品。
1.2 实验装置与流程
实验装置由配气系统、反应系统和检测系统3部分组成,采用固定床催化脱硝反应器测定不同催化剂样品的脱硝效率。配气系统所用气源为1%NO/N2气瓶气、1%NH3/N2气瓶气、高纯O2(99.999%)和高纯N2(99.999%)。模拟烟气由500×10−6NO,500×10−6NH3,3.4%O2和N2(载气)组成(均为质量分数),各种气体由质量流量计精确控制,气体总流量300 mL/min,催化剂用量0.5 g,空速为24 000 h−1。反应器为内径10 mm的石英玻璃管,置于管式电炉内,反应温度由温控仪精确控制,催化剂放置在反应器中间区域的石英沙芯上。采用Kane KM9106烟气分析仪测量进出口NO等气体的浓度。实验流程如图1所示。
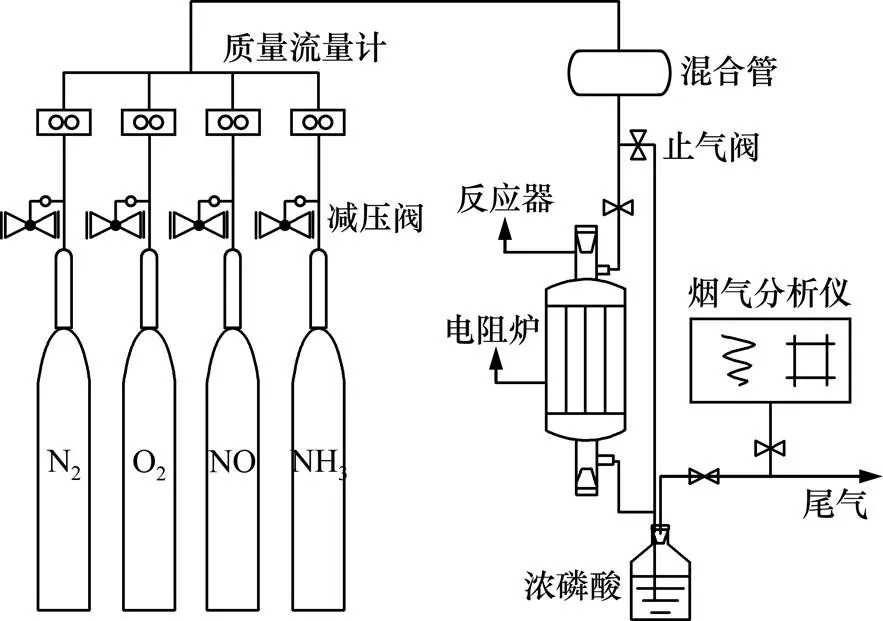
图1 反应流程示意图
1.3 活性及再生效果评价
以NO转化率作为催化剂催化性能指标,计算式为
其中:0和分别表示测定温度下的反应器进出口NO的稳定质量分数(10−6)。
催化剂再生效果以再生催化剂NO转化率(2)与失活催化剂NO转化率(1)的差值和新鲜催化剂NO转化率(0)与失活催化剂NO转化率(1)的差值的比值作为评价指标,计算式为
1.4 样品表征
采用德国ZEISS EVO 18型电子扫描显微镜分析催化剂形貌和表面形态;美国麦克仪器公司ASAP 2020全自动快速比表面积及介孔/微孔分析仪测定催化剂比表面积和孔结构特性;英国岛津/Kratos Amicus X射线光电子能谱仪对催化剂表面化学结构进行分析。
2 结果与讨论
2.1 催化剂的脱硝效率
图2所示为不同温度下(200~500 ℃)新鲜催化剂、失活催化剂及水洗和酸洗再生催化剂的表观活性。新鲜催化剂具有较宽的活性温度窗口和较高的催化脱硝性能,在250~450 ℃的温度范围内表现出很好的催化活性(>97%),250 ℃即可达到97%的NO转化率,400 ℃时,NO转化率高达99%,且反应温度升至500℃时,新鲜催化剂的脱硝效率为87%。而失活催化剂在各测试温度下的脱硝效率均明显降低,脱硝效率不足20%,且随着反应温度的增加,其高温催化活性逐渐降低。

1—新鲜催化剂;2—水洗再生催化剂;3—酸洗再生催化剂;4—失活催化剂
通过水洗和H2SO4溶液洗涤对失活催化剂进行再生,并计算得其再生后的脱硝效率。由图2可见:2种再生方法均能使失活催化剂的部分脱硝性能得到恢复,整体来看,酸洗再生效果较水洗再生效果好,然而,再生后的最佳活性温度窗口均变窄,可能是由于再生过程中,催化剂表面的Na2O得以去除但不完全,同时催化剂表面的活性组分也可能流失。值得注意的是,水洗再生和酸洗再生后的催化剂活性在低温段(温度<350 ℃)存在明显的差异,这是因为硫酸洗液较水溶液相比,对失活催化剂表面的碱金属物种具有更好的溶解作用,催化剂经过硫酸清洗后,去除大量碱金属物种的同时,催化剂表面可能硫酸化,且形成的SO42−活性基团对SCR反应也具有一定的促进作用。
以式(2)作为催化剂再生效果的评价指标,考察不同方法对失活催化剂的再生能力,计算结果如表1所示,并结合图2对水洗和酸洗再生效果进行分析。在反应温度<350 ℃时,水洗再生催化剂的催化活性不高,即水洗再生效果不是很好,且明显低于0.5 mol/L H2SO4洗涤的再生效果(350 ℃的脱硝率达到98%,再生效果高达99.98%),反应温度在350~500 ℃范围内,水洗再生效果较突出(再生效果>95%)。
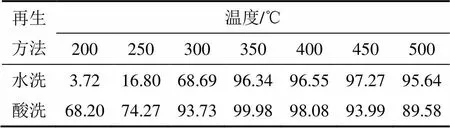
表1 水洗和酸洗对失活催化剂的再生效果(质量分数)
2.2 SEM分析
图3所示为催化剂的SEM像。由图3(a)可知:新鲜催化剂的颗粒比较均一,分布比较均匀,未出现团聚和板结现象。而失活催化剂颗粒的表面形貌发生了变化,出现板结和团聚的现象(图3(b)),可能是由于Na2O晶体覆盖催化剂表面并引起其孔道堵塞。从图3(c)可以看出:水洗再生后的催化剂部分表面仍存在板结和团聚现象,但较失活催化剂样品的有所减弱,且表面颗粒状物质减少,可能是催化剂表面覆盖的Na2O经水洗后脱落。值得注意的是,图3(d)所示的酸洗催化剂颗粒表面与其他3种催化剂表面相比,粗糙度明显降低,且有些许大颗粒物质出现,说明在酸洗过程中,催化剂表面可能生成了硫酸盐,并引起催化剂表面硫酸化。

(a) 新鲜催化剂;(b) 失活催化剂;(c) 水洗再生催化剂;(d) 酸洗再生催化剂
2.3 BET分析
为了研究催化剂失活及再生后其比表面积和孔结构特性的变化,对各催化剂样品进行了BET测试。图4所示为各催化剂样品的吸附−脱附等温线图及孔径分布图。从图4可以发现:4种催化剂样品等温线类型皆为Ⅳ型等温线,并拥有H1型滞后环,并且滞后环结束位置均在相对压力为0.7左右,但失活催化剂的吸附量有所降低,且经水洗和酸洗再生后的催化剂的吸附量得到不同程度的回升,尤其是酸洗再生后催化剂的吸附量比新鲜催化剂的吸附量大。从图4还可以看出:新鲜、失活及水洗和酸洗再生后的催化剂的孔径分布并未发生很大的变化。
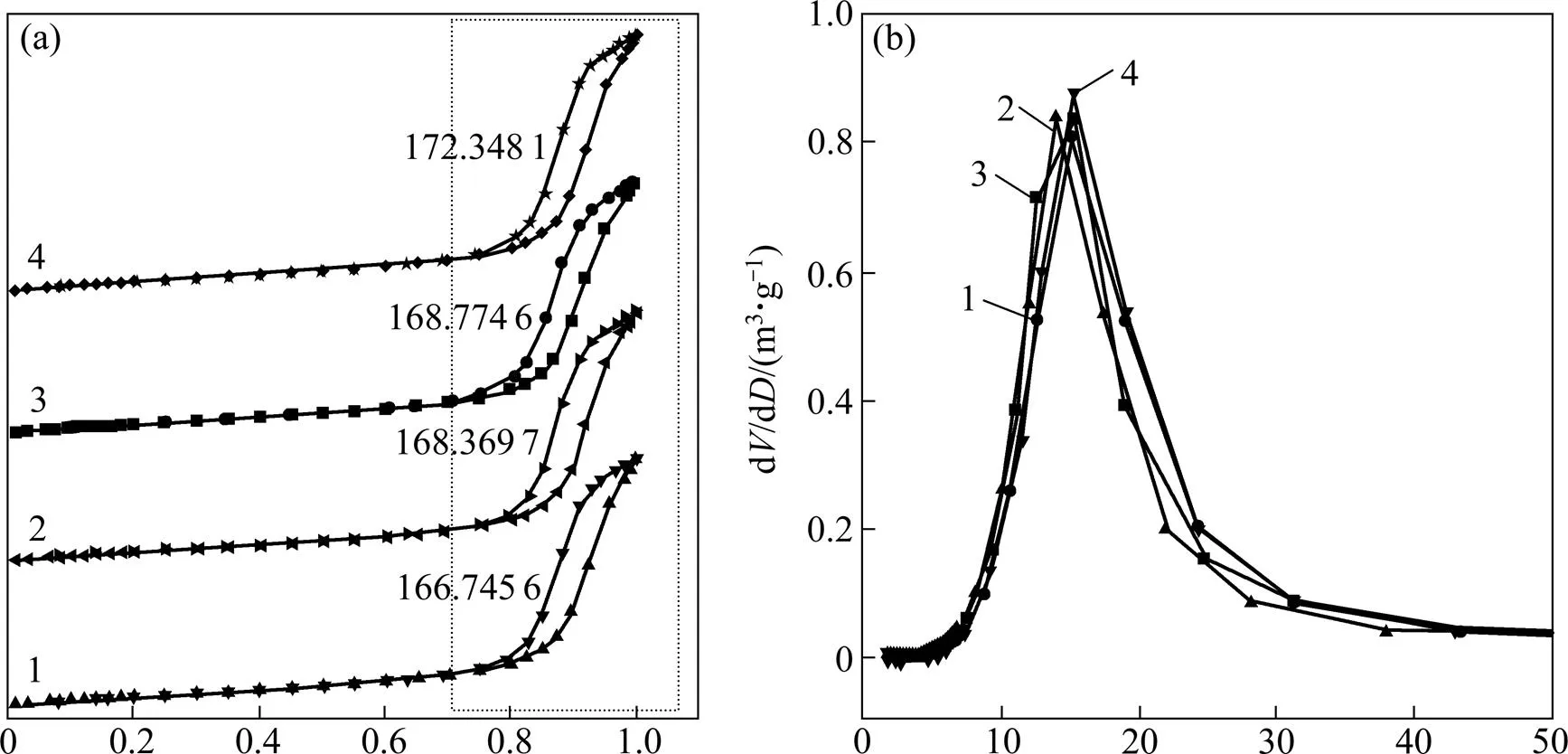
1—失活催化剂;2—水洗再生催化剂;3—新鲜催化剂;4—酸洗再生催化剂
表2列出了新鲜、失活、再生后催化剂的比表面积、平均粒径和总孔体积的对比。由表2可知:较新鲜催化剂相比,失活催化剂的比表面积和总孔体积均略微减小,而平均孔径略微增大,但这些变化是微弱的。另外,经水洗和酸洗再生后的催化剂的比表面积和孔结构都有不同程度的恢复,尤其是酸洗再生后的催化剂的比表面积比新鲜催化剂的大了约1.5 m2/g,可能是由于酸洗后催化剂表面硫酸化引起的。
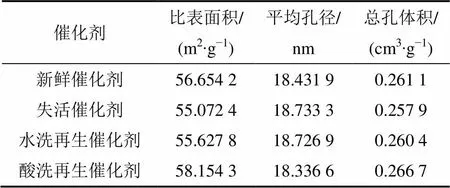
表2 催化剂的比表面积和孔结构参数
结合图3,4和表2分析认为:催化剂表面盐颗粒的沉积和催化剂部分孔洞的堵塞,引起催化剂的比表面积、平均粒径和总孔体积发生微弱的变化,但这些微弱的变化不足以引起催化剂显著的活性变化。在较低含量的碱金属引起催化剂中毒的机理中,物理中毒不是催化剂失活的主要原因。
2.4 XPS分析
为进一步研究碱金属钠的中毒对催化剂表面性质的影响,分别对新鲜催化剂、钠中毒催化剂及水洗和酸洗再生后的催化剂进行XPS表征和分析。各催化剂样品的XPS数据如表3所示。由表3可知:催化剂的主要成分为Ti和O,约占77%(质量分数,下同),活性组分V和W分别约占1%和8%左右。经钠盐处理后的催化剂样品的Na负载量约为6.74%,结合图2失活催化剂的脱硝效果可以看出,这样的负载量足以使新鲜催化剂失去大部分脱硝活性。从表3可以看出:经中毒处理后的失活催化剂表面如Ti,O和V的含量均减少,且元素O的损失量较大,由原来的37.89%将至34.99%,而作为活性组分之一的W元素的含量由7.53%增加到11.66%。经水洗和酸洗再生后的催化剂表面Na元素减少,水洗仅去除了约33.98%的Na元素,而酸洗去除了催化剂表面大部分的Na(85.90%)。另外,经2种再生方法均可恢复催化剂表面的Ti和O元素的含量,而较失活催化剂表面的V和W元素含量相比,经水洗处理后的失活催化剂表面的V和W元素的含量几乎没有发生变化,而经酸洗处理后的失活催化剂表面V元素的流失量较大(约47%)。但酸洗对失活催化剂的再生效果很好,在200~500 ℃的温度范围内表现出较好的脱硝性能(见图2)。
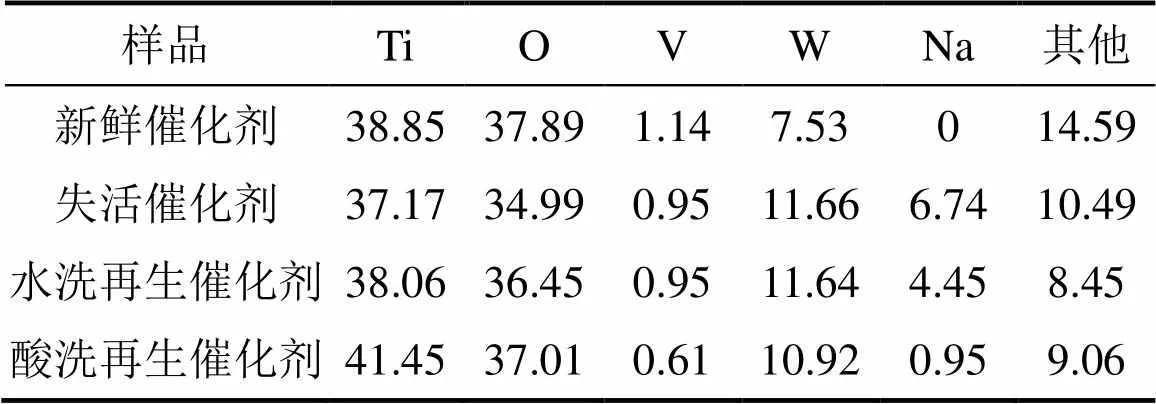
表3 新鲜/失活/再生催化剂的XPS分析结果(质量分数)
值得注意的是,中毒后的催化剂表面O元素大量减少,而经水洗和酸洗再生后的催化剂表面O含量基本恢复,分别为新鲜催化剂表面O含量的96%和98%。Jing等[13]发现,催化剂表面的化学吸附氧是最活跃的氧物种,在氧化反应中具有重要的作用。Chen等[14]研究发现,K2O和Na2O导致催化剂表面化学吸附氧的数量减少以及钒物种和钨物种还原能力下降,这是导致催化剂脱硝活性降低的主要原因之一。
图5所示为催化剂的O1s谱。据XPS谱图手册和相关文献记载[15−16],结合能=528~531 eV归属于催化剂表面的金属晶格氧,=532 eV归属于催化剂表面的化学吸附氧。由图5(a)可以看出:新鲜催化剂表面结合能为530.5 eV和529.9 eV处的氧均属于V2O5-WO3/TiO2金属氧化物体系中的晶格氧,而结合能为532 eV属于催化剂表面的化学吸附氧。催化剂Na中毒后,其表面金属晶格氧的结合能向低结合能处偏移(Δ0.5 eV,0.4 eV),表明Na的引入改变了催化剂的表面性能,使V2O5和WO3等金属氧化物的化学环境发生变化,从而影响其催化性能。水洗和酸洗再生后的催化剂表面晶格氧的结合能逐渐向高结合能处偏移,其中酸洗后催化剂表面晶格氧的结合能的位置与新鲜催化剂的基本一致。由图5可以看出:失活催化剂表面化学吸附氧的含量大量减少,水洗再生后的催化剂表面的化学吸附氧的含量增加,然而,酸洗再生后的催化剂表面化学吸附氧消失,值得注意的是,在=532.5 eV处出现微弱的峰(图5(d)),归属于SO42−中的氧[17],进一步说明经酸洗后催化剂表面有硫酸盐存在,即证实了前面有关酸洗引起催化剂表面硫酸化的推测。此结论与Kang等[18]的研究相吻合,即化学吸附氧占催化剂表面氧物种的比例与催化剂表面催化活性有一定的正相关性,即化学吸附氧含量越高,越有利于SCR的反应。
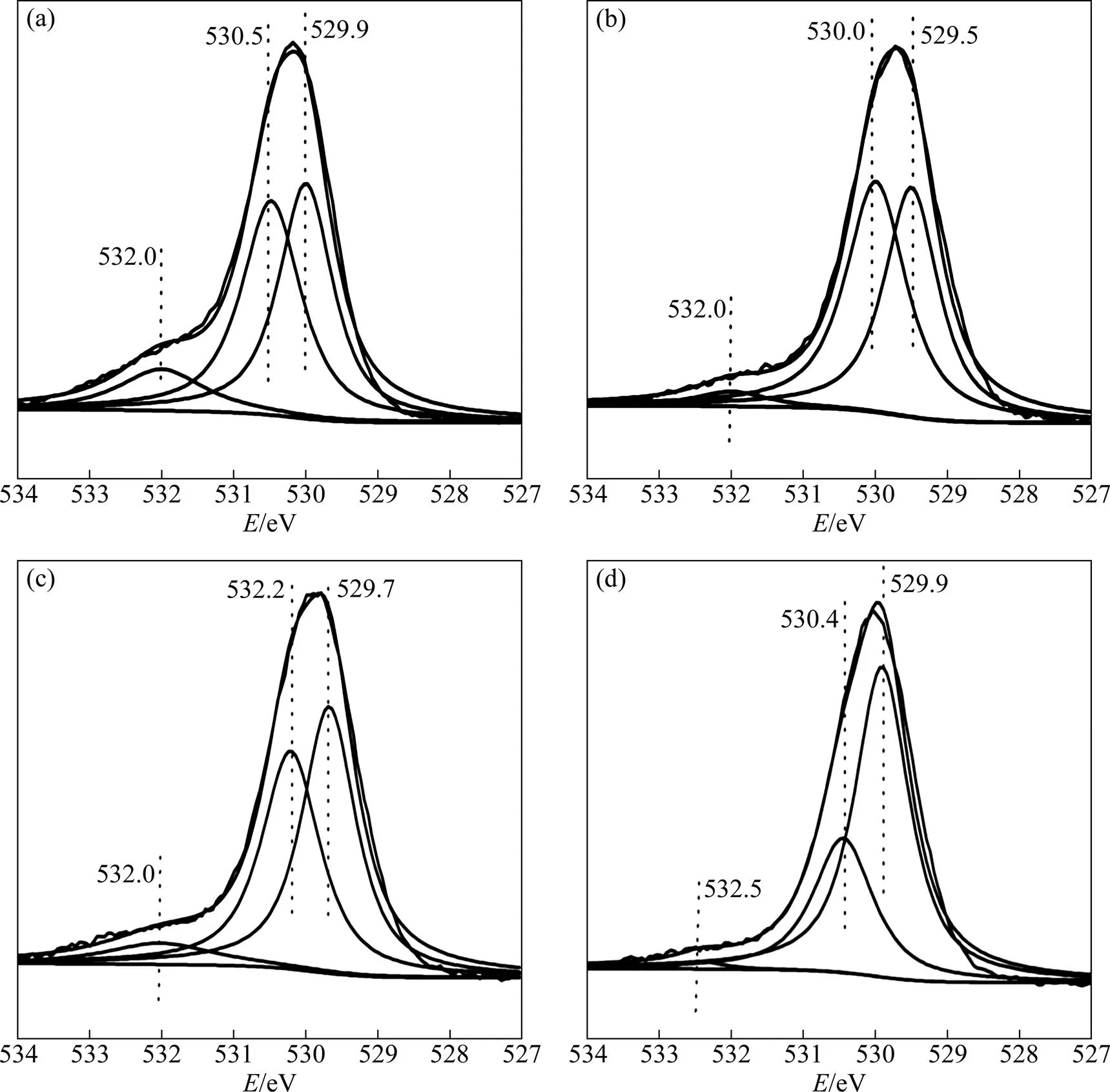
(a) 新鲜催化剂;(b) 失活催化剂;(c) 水洗再生催化剂;(d) 酸洗再生催化剂
2.5 SCR反应及催化剂失活机理分析
V2O5/TiO2催化剂中的TiO2载体仅呈现L酸性,V2O5也只有L酸性位,吸水后可以转变为B酸性位,如图6所示[19]。在NH3-SCR反应中,NH3通常以NH3分子和NH4+离子分别吸附在V2O5/TiO2催化剂表面的L酸性位和B酸性位[20],根据NH3吸附位不同,SCR反应机理分为L酸机理[19]和B酸机理[21],其中支持B酸机理的研究者比较多。
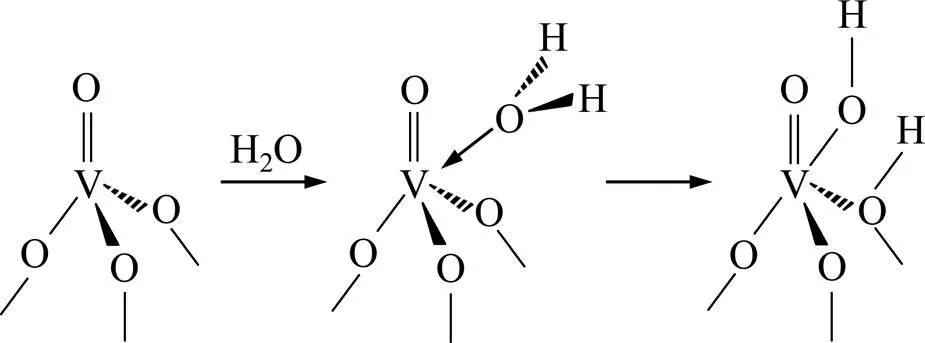
图6 表面钒中心的L酸性位向B酸性位转变示意图
TopsØe等[22]研究表明,B酸性位上的NH4+在SCR反应中起主要作用,V—OH中的V以五价氧化态即V5+—OH的形式存在,并提出了在有氧条件下V2O5/TiO2催化剂上的SCR反应历程,如图7所示。NH3吸附在B酸性位上(V5+—OH)形成—NH4+,并被邻近的V5+=O氧化形成—NH3+,而V5+=O被还原为V4+—OH,—NH3+与NO结合形成—NH3+—NO并分解成N2和H2O,V4+—OH在O2的作用下氧化为V5+=O。 TopsØe认为NH3-SCR反应涉及到2个V位,分别是吸附位V5+—OH和氧化位V5+=O,吸附在B酸性位上的—NH4+脱氢形成—NH3+是反应的关键步骤。
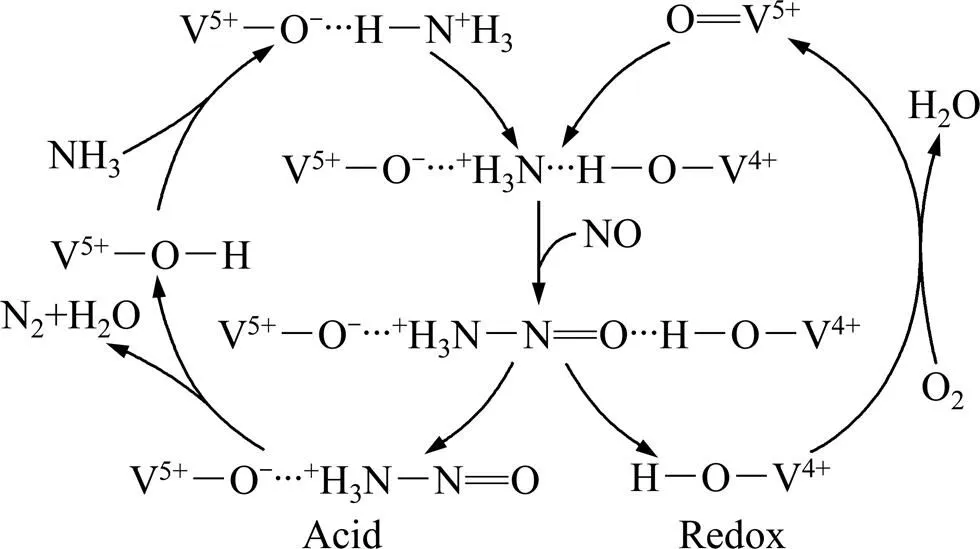
图7 有氧条件下V2O5/TiO2催化剂上的SCR反应历程
基于V2O5/TiO2催化剂上的SCR反应机理,Zheng等[23]对碱金属引起催化剂化学中毒进行分析:碱金属(K,Na)与催化剂表面的B酸性位点上的V—OH发生反应,生成V—OK或V—ONa,使催化剂吸附NH3的能力下降,从而使参与NO还原反应的NH3的吸附量减少,并降低了其参与SCR反应的活性。同时研究表明,催化剂碱中毒失活速率远大于比表面积减少的速率,由此可知,碱金属引起的催化剂中毒以化学中毒为主。
结合本研究的相关结果及对钒钛系催化剂表面氧物种的分析(图5),失活催化剂表面金属晶格氧的结合能的偏移,表明Na的引入改变了催化剂的表面性能,使V2O5和WO3等金属氧化物的化学环境发生变化。由此我们推测,经钠盐处理后的催化剂表面活性位点上形成了V—ONa,引起催化剂化学中毒,从而影响其催化性能。
根据TopsØe等提出的有氧条件下钒钛系催化剂上的SCR反应历程(图7),V5+=O被还原为V4+—OH及V4+—OH再氧化为V5+=O的过程也是整个NH3-SCR反应的关键步骤。由此可知,催化剂表面V的价态对其催化性能也有一定的影响。图8所示为催化剂的V2p3/2谱。由图8可知,失活催化剂表面V的结合能较新鲜催化剂的有所偏移,因此可以判断,Na的引入改变了V的化学环境,结合B酸机理和O1s谱图的分析,进一步证实了催化剂碱中毒的化学机理,即Na与催化剂表面的V—OH反应,形成了V—O—Na。

(a) 新鲜催化剂;(b) 失活催化剂;(c) 水洗再生催化剂;(d) 酸洗再生催化剂
由图8可以发现:催化剂表面V4+和V5+所占的比例((V4+)/(V5+))发生了变化。表4列出了不同催化剂表面V4+和V5+的含量及其比例。失活催化剂表面(V4+)/(V5+)较新鲜催化剂表面(V4+)/(V5+)大幅度降低,由1.43减小到0.69,减少了约52%。同时,经过水洗和酸洗再生后的催化剂表面(V4+)/(V5+)不断增加,分别为0.82和0.96,仅为新鲜催化剂表面(V4+)/(V5+)的57%和67%。由此,我们认为,催化剂的表面活性不但与其酸性位点、表面化学吸附氧的含量有关,而且还与其表面活性中心上V的形态及(V4+)/(V5+)存在一定的正相关性。此结论与Shang等[24]的研究结果相类似。

表4 新鲜/失活/再生催化剂表面V的价态
3 结论
1) 较低负载量的Na2O可以使V2O5/TiO2基SCR催化剂严重失活,水洗和酸洗再生均能使失活催化剂的大部分脱硝性能得到恢复,整体来看,酸洗再生效果较水洗再生效果好,且催化剂表面硫酸化有利于催化反应的进行。
2) 碱金属钠盐可引起催化剂物理中毒和化学中毒,其中以化学中毒为主。物理中毒主要是引起催化剂表面颗粒的沉积和孔道的堵塞。而化学中毒主要是因为碱金属Na与催化剂表面的B酸性位点上的V-OH发生反应,生成V-ONa,使V2O5和WO3等金属氧化物的化学环境发生变化,从而影响其催化性能。
3) V2O5/TiO2催化剂表面酸性位点为Brönsted酸,碱中毒以及再生不会影响催化剂表面酸位点类型。
4) 催化剂的脱硝活性与其表面的化学吸附氧量和表面活性中心上(V4+)/(V5+)的比有一定的关联性。
[1] Hao JM, Tian H Z, Lu Y Q. Emission inventories of NOfrom commercial energy consumption in China, 1995-1998[J]. EnvironmentalScience andTechnology, 2002, 36(4): 552−560.
[2] Roy S, Hegde M S, Madras G. Catalysis for NO abatement[J]. Applied Energy, 2009, 86(11): 2283−2297.
[3] Amiridis M D, Duevel R V, Wachs I E. The effect of metal oxide additives on the activity of V2O5/TiO2catalysts for the selective catalytic reduction of nitric oxide by ammonia[J]. Applied Catalysis B:Environmental, 1999, 20(2): 111−122.
[4] 云端, 宋蔷, 姚强. V2O5-WO3/TiO2SC R催化剂的失活机理及分析[J]. 煤炭转化, 2009, 32(1): 91−96.
YUN Duan, SONG Qiang, YAO Qiang. Mechanism and analysis of SCR catalyst deactivation[J]. Coal Conversion, 2009, 32(1): 91−96.
[5] 商雪松, 陈进生, 赵金平, 等. SCR脱硝催化剂失活及其原因研究[J]. 燃料化学学报, 2011, 39(6): 465−471.
SHANG Xuesong, CHEN Jinsheng, ZHAO Jinping, et al. Discussion on the deactivation of SCR denitrification catalyst and its reasons[J]. Journal of Fuel Chemistry and Technology, 2011, 39(6): 465−471.
[6] 姜烨, 高翔, 吴卫红, 等. 选择性催化还原脱硝催化剂失活研究综述[J].中国电机工程学报, 2013, 33(14): 18−31. JIANG Ye, GAO Xiang, WU Weihong, et al. Review of the deactivation of selective catalytic reduction DeNOx catalysts[J]. Proceedings of the CSEE, 2013, 33(14): 18−31.
[7] Kamata H, Takahashi K, Odenbrand C U I. The role of K2O in the selective reduction of NO with NH3over a V2O5(WO3)/TiO2commercial selective catalytic reduction catalyst[J]. Journal of Molecular Catalysis A, Chemical, 1999, 139(2/3): 189−198.
[8] Francesco C, Anker D J, Jan E J, Rasmus F. Influence of reaction products of K-getter fuel additives on commercial vanadia-based SCR catalysts, Part I. Potassium phosphate[J]. Applied Catalysis B: Environmental, 2009, 86(3/4): 196−205.
[9] Benson S A, Laumb J D, Crocker C R, et al. SCR Catalyst Performance in Flue Gases Derived from Subbituminous and Lignite Coals[J]. Fuel Processing Technology, 2005, 86(5): 577−613.
[10] 朱崇兵, 金保升, 仲兆平, 等. K2O对V2O5-WO3/TiO2催化剂的中毒作用[J]. 东南大学学报(自然科学版), 2008, 38(1): 101−105.
ZHU Chongbing, JIN Baosheng, ZHONG Zhaoping, et al. Poisoning effect of K2O on V2O5-WO3/TiO2catalyst[J]. Journal of Southeast University (Natural Science Edition). 2008, 38(1): 101−105.
[11] Chen J P, Yang R T. Mechanism of poisoning of the V2O5/TiO2catalyst for the reduction of NO by NH3[J]. Journal of Catalysis, 1990, 125(2): 411−420.
[12] 沈伯雄, 施建伟, 杨婷婷, 等. 选择性催化还原脱氮催化剂的再生及其应用评述[J]. 化工进展, 2008, 27(1): 64−67.
SHEN Boxiong, SHI Jianwei, YANG Tingting, et al. Regeneration technologies of SCR catalysts and their applications[J]. Chemical Industry and Engineering Progress, 2008, 27(1): 64−67.
[13] Jing L Q, Xu Z L, Sun X J, Shang J, Cai W M. The surface properties and photo-catalytic activities of ZnO ultrafine particles[J]. Applied Surface Science, 2001, 180(3/4): 308−314.
[14] Chen L, Li J H, Ge M F. The poisoning effect of alkali metals doping over nano V2O5-WO3/TiO2catalysts on selective catalytic reduction of NOby NH3[J]. Chemical Engineering Journal, 2011, 170(2/3): 531−537.
[15] Hardacre C, Roe G W, Lambert R M. Structure, composition and thermal properties of cerium oxide films on platinum {111}[J]. Surface Science, 1995, 326(1/2): 1−10.
[16] Tam K H, Cheung C K, Leung Y H, Djurišić A B, Ling C D, Beling C D, Fung S, Kwok W M, Chan W K, Philips D L, Ding L, Ge W K. Defects in ZnO Nanorods Prepared by a Hydrothermal Method[J]. The Journal of Physical Chemistry B, 2006, 110(42): 20865−20871.
[17] Ebitani K, Konno H, Tanaka T, Hattori. In-situ XPS study of zirconium oxide promoted by platinum and sulfate ion[J]. Journal of Catalysis, 1992, 135(1): 60−67.
[18] Kang M, Park E D, Kim J M, Yie J E. Manganese oxide catalysts for NOreduc-tion with NH3at low temperatures[J]. Applied catalysis A: General, 2007, 327(2): 261−269.
[19] Ramis G, Busca G, Lorenzelli V, Forzatti P. Fourier transform infrared study of the adsorption and coadsorption of nitric oxide, nitrogen dioxide and ammonia on TiO2anatase[J]. Applied Catalysis, 1990, 64(12): 243−257.
[20] Busca G, Lietti L,Ramis G, Berti F. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: A review[J]. Applied Catalysis B:Environmental, 1998, 18(1/2): 1−36.
[21] TopsØe N Y, TopsØe H, Dumesic J A. Vanadia/Titania Catalysts for Selective Catalytic Reduction (SCR) of Nitric-Oxide by Ammonia: I. Combined Temperature-Programmed in-Situ FTIR and On-line Mass-Spectroscopy Studies[J]. Journal of Catalysis 1995, 151(1): 226−240.
[22] TopsØe N Y, Dumesic J A, TopsØe H. Vanadia-Titania Catalysts for Selective Catalytic Reduction of Nitric-Oxide by Ammonia: Ⅱ. Studies of Active Sites and Formulation of Catalytic Cycles[J]. Journal of Catalysis, 1995, 151(1): 241−252.
[23] Zheng Y, Jensen A D, Johnsson J E. Laboratory investigation of selective catalytic reduction catalysts: Deactivation by potassium compounds and catalyst generation[J]. Industrial & Engineering Chemistry Research, 2004, 43(4): 941−947.
[24] Shang X S, Hu G R, He C, Zhao J P, Zhang F W, Xu Y, Zhang Y F, Li J R, Chen J S. Regeneration of full-scale commercial honeycomb monolith catalyst (V2O5-WO3/TiO2) used in coal-fired power plant[J]. Journal of Industrial and Engineering Chemistry, 2012, 18(1): 513−519.
(编辑 陈爱华)
Sodium poisoning mechanism and regeneration of commercial De-NOSCR catalysts
GAO Fengyu, TANG Xiaolong, YI Honghong, ZHAO Shunzheng, LI Dong, MA Ding, ZHANG Tongtong
(Civil and Environmental Engineering School, University of Science and Technology Beijing, Beijing 100083, China)
Sodium poisoning of commercial De-NOSCR catalysts (V2O5-WO3/TiO2) and the regeneration of deactivated catalysts by water and 0.5 mol/L H2SO4were studied under simulated condition in laboratory. The physicochemical properties of catalysts were characterized by SEM, BET and XPS measurements, the reaction and deactivation mechanism of V2O5/TiO2were also analyzed and discussed. The results indicate that catalysts are poisoned seriously with low-capacity Na2O, the catalytic activity of deactivated catalysts are recovered at varying degrees by washing with water and sulfuric acid, and the regeneration effect of washing with sulfuric acid is better than that of washing with water. Deposition of sodium salts particles and blocking of the pores and channels cause the physical poisoning of catalysts, and the main reason of chemical poisoning is attributed to the reaction of Na and V—OH to V—O—Na on the Brönsted acid sites, which results in the changes of the chemical environment of metallicoxide on the surface of catalysts, thus blocking the spread of NO and NH3to the inner of catalysts. The results also indicate that the denitration performance of SCR catalysts has certain positive correlation with the contents of chemical adsorbed oxygen and the ratio of(V4+)/(V5+) on the surface of catalysts.
commercial V2O5/TiO2catalysts; sodium poisoning;regeneration; reaction mechanism; deactivated mechanism
10.11817/j.issn.1672-7207.2015.06.052
X511;O643.32+2
A
1672−7207(2015)06−2382−09
2014−04−13;
2014−07−20
国家自然科学基金资助项目(21177051);新世纪优秀人才支持计划(NECT-13-0667);首都蓝天行动培育专项课题(Z141100001014006);中央高校基本科研业务费专项资金(06101047)(Projects (21177051) supported by the National Natural Science Foundation of China; Project (NECT-13-0667) supported by Program for New Century Excellent Talents in University, Project (Z141100001014006) supported by The Special Project on Air Pollution Control of Beijing Municipal Science & Technology Commission; Project (06101047) supported by The Fundamental Research Funds for the Central Universities)
唐晓龙,教授,博导,从事环境污染控制与治理、环境功能材料等研究;电话:010-62332747;E-mail:txiaolong@126.com
