PPARα/COX-2/PGE2通路在人结肠癌耐药细胞中的作用研究
2015-09-26赵贤元余跃天李昱洁曹建国
聂 芳 赵贤元 余跃天 殷 荣 李昱洁 曹建国 皋 源
PPARα/COX-2/PGE2通路在人结肠癌耐药细胞中的作用研究
聂芳赵贤元余跃天殷荣李昱洁曹建国皋源
目的初步检测PPARα对COX-2蛋白表达和酶活性的影响,推测PPARα/COX-2/PGE2通路在人结肠癌耐药细胞中的可能作用。方法Real-Time PCR和Western blot检测SW1116细胞、SW1116/HCPT细胞,以及下调PPARα表达后SW1116细胞中COX-2的表达差异;ELISA检测WY-14643分别干预SW1116细胞,SW1116/HCPT细胞,以及下调PPARα表达后各组细胞培养上清中PGE2含量;流式细胞术检测不同浓度PGE2对细胞周期的影响。结果COX-2在耐药细胞中表达明显下调。PPARα表达下调抑制COX-2表达。PPARα激动剂WY-14643促进细胞释放PGE2[250 μM:(0.7851± 0.0203)ng/mL vs(0.2443±0.01228)ng/mL,P=0.000;100 μM:(0.5483±0.0614)ng/mL vs(0.2443± 0.01228)ng/mL,P=0.0083];PGE2降低细胞周期G1期比例,升高S期比例,PI由对照组的(32.2± 2.13)%,上升到(42.2±3.14)%、(52.1±2.43)%,P=0.0123。结论人结肠癌耐药细胞中COX-2表达显著下调;PPARα有促进COX-2蛋白表达和酶活性的作用。
结直肠癌;环氧合酶2;耐药;PGE2;PPARα
【Abstract】Objective To explore the regulation of PPARα to COX-2 protein expression and COX-2 enzyme activity,and to find out the role of PPARα/COX-2/PGE2pathway in drug resistance colorectal cancer cells.Methods The expressions of COX-2 were measured by Realtime PCR and Western blot in SW1116 cells,SW1116/HCPT cells and SW1116 cells transfected with siRNA vectors for silencing PPARα;PGE2concentration in culture supernatants was detected by ELISA in SW1116 cells and SW1116/HCPT cells treated with PPARα agonist WY-14643,SW1116 cells and SW1116/HCPT cells transfected with siRNA vectors for silencing PPARα;cell cycles of SW1116/HCPT cells treated with PGE2were assessed by flow cytometry.Results Protein and mRNA expressions of COX-2 were much lower in SW1116/HCPT cells and SW1116 cells transfected with siRNA vectors for silencing PPARα than in SW1116 cells.PGE2concentrations in cells culture supernatants of SW1116/HCPT cells were elevated after treatment with WY-14643[100 μM:(0.5483± 0.0614)ng/mL;250 μM:(0.7851±0.0203)ng/mL];proliferation index were elevated in SW1116/HCPT cells with PGE2[10-8mol/L:(42.2±3.14);10-7mol/L:(52.1±2.43)%;con:(32.2±2.13)%].Conclusions The expression of COX-2 were much lower in SW1116/HCPT cells than in SW1116 cells.PPARα promoted the protein expression and enzyme activity of COX-2.
【Key words】Colorectal Cancer;COX-2;Drug Resistance;PGE2;PPARα
环氧合酶-2(cyclooxygenase-2,COX-2)作为前列腺素合成的限速酶在炎症中起重要作用,而且参与结直肠癌发生、发展及促进其转移与侵袭。COX-2主要表达在新生的血管内皮细胞和形成血管的细胞中,通过影响VEGF参与肿瘤新生血管的形成[1-2]。在人结肠癌羟基喜树碱(Hydroxycamp-tothecin,HCPT)多药耐药细胞株(简称SW1116/ HCPT)中,过氧化物酶体增殖物激活受体α(Peroxisome proliferator-activated receptors-α,PPARα)下调了105倍[3]。进一步研究发现,COX-2的表达在该耐药细胞中也是明显下调的。为了明确PPARα、COX-2在结肠癌细胞中的作用,探讨可能相关的信号通路的效应分子,我们假定PPARα可能在某种程度上发挥对COX-2调节作用,并以此为基础,通过检测COX-2的作用底物PGE2,了解PPARα激动剂WY-14643对COX-2的活性和蛋白表达影响,进一步推测PPARα对COX-2调节的可能机制。
材料和方法
一、材料
1.细胞株和干扰质粒
人结肠癌SW1116细胞株,人结肠癌羟基喜树碱多药耐药SW1116/HCPT细胞株(简称为SW1116/HCPT),由仁济医院消化科冉志华教授提供,该多药耐药细胞株是通过药物浓度梯度法诱导建立[3],该细胞株冻存前培养于含羟基喜树碱(浓度为1 μg/mL)的RPMI-1640完全培养液,复苏后培养于无羟基喜树碱的RPMI-1640完全培养液3~5代后用于实验。PPARa干扰质粒由课题组童锦禄博士构建完成并保存,载体为p-RNAT-U6.1/neo[4]。
2.主要试剂
脂质体2000(美国Invitrogen公司);羟基喜树碱(HCPT):长春天诚药业有限公司;ELISA-PGE2试剂盒:美国R&D公司(货号KGE004);PGE2:美国SIGMA公司(货号P0409);WY-14643:(美国SIGMA);MK-886:(美国SIGEMA);RKIP:兔抗人多克隆抗体(Santa Cruz,货号sc-28837);COX-2:山羊抗人多克隆抗体(美国Abcam,货号ab23672);PPARα:鼠抗人单克隆抗体(美国Abcam公司,货号ab2779);HRP标记的小鼠抗人GAPDH多克隆抗体(上海康成生物,货号KC-5G5)。
二、主要方法
1.实时荧光定量PCR
(1)Trizol法分别抽提细胞RNA:SW1116细胞和人结肠癌羟基喜树碱多药耐药细胞SW1116/ HCPT细胞,每孔3×105接种于六孔板,待细胞生长汇度达85%~95%时,Trizol法分别抽提两组细胞RNA,紫外分光光度法检测RNA浓度分别为1.41 μg/uL、1.35 μg/uL;并用变性琼脂糖凝胶电泳鉴定总RNA分子完整性。
(2)逆转录PCR合成cDNA。在RNA酶灭活的0.2 mL PCR管中配置20 μL如下反应体系:5× PrimeScripBuffer 2.0 μL,PrimeScripRT Enzyme MixI(10 mM)0.5 μL,Oligo dT Primer,Random primer各0.5 μL,总RNA≤1.0 μg,最后加入RNase free H2O至20 μL;37℃15 min--85℃5 s --4℃维持,得到RT反应液,即cDNA模版。
(3)Real-Time PCR。配置20 μL如下反应体系:SYBRPremix Ex TaqTMI(I2×)10.0 μL,Forward Prime(r10 μM)0.8 μL,Reverse Prime(r10 μM)0.8 μL,ROX Reference Dye 0.4 μL,RT反应液≤2.0 μL,最后加RNase free H2O至20 μL,反应条件:95℃30 s--95℃5 s,60℃31 s重复40个循环--72℃45 s。

(4)结果计算:每个样品进行3次重复试验。记录各孔的Ct值。每个样品的Ct值以3个复孔的平均Ct值±标准差(±s)表示。采用经典的2-ΔΔCt相对定量法计算:△Ct(目的基因)=目的基因Ct值-内参对照基因的Ct值,△△Ct=△Ct(目的基因)-△Ct(标准值),目的基因的相对总量为2-ΔΔCt。Ct值是每个反应管内的荧光信号到达设定阈值时所经历的循环数,Ct值与miRNA的表达水平呈负相关,即CT值越高,说明miRNA表达水平越低。
2.Western blot检测COX-2的蛋白表达
(1)实验分组:细胞均接种于六孔板,3×105/孔。①SW1116细胞,分别转染阴性对照质粒和PPARα干扰质粒,转染72 h后检测PPARα干扰质粒对PPARα蛋白水平的抑制情况;②人结肠癌羟基喜树碱多药耐药SW1116/HCPT细胞株,每孔加入培养液2 mL,12 h后予以PPARα激动剂WY-14643进行干预,终浓度分别为50 μM、100 μM、250 μM,阴性对照为同等浓度的DMSO加培养液,干预48 h后,抽提蛋白;③SW1116细胞和人结肠癌羟基喜树碱多药耐药SW1116/HCPT细胞,分别收集细胞总蛋白。
(2)实验方法:用含有蛋白酶抑制剂的细胞裂解液细胞,抽提细胞总蛋白,Bradford法进行蛋白定量,配置10%分离胶,4%的基层胶,上样,每孔加入50 μg蛋白,进行SDS-PAGE电泳,PVDF转膜封闭,加入一抗,二抗孵育,洗膜,化学发光;同上操作进行内参照GAPDH检测,ImageJ 1.5软件分析条带灰度值。
3.ELISA检测细胞培养上清中PGE2含量
(1)实验分组:同western blot,细胞接种于六孔板,3×105/孔,每孔最终培养液体积为1.5 mL。①转染后24 h,48 h后收集细胞培养上清;②WY-14643进行不同浓度干预,作用24 h后收集细胞培养上清;③接种48 h后收集细胞培养上清。
(2)制备PGE2不同浓度标准品,工作液及待测样品:①按浓度梯度稀释法,配置浓度分别为2 500 pg/mL、1 000 pg/mL、500 pg/mL、100 pg/mL、50 pg/ mL的PGE2工作液。配置好的PGE2工作液在60 min内进行检测。②取20 mL洗脱液,加入双蒸水,1:25倍稀释后放于4℃备用。③将过氧化氢和四甲基联苯胺等体积混匀,配置成底物反应溶液,15 min内避光备用。
(3)收集细胞培养上清:取0.5 mL培养上清,加入0.5 mL 800 mol/L乙醇及10 μL冰醋酸轻微混合,室温静置5 min,以4 000 g离心10 min,清除培养液中残留的颗粒和细胞,取上清放入1.5 mL无菌EP管中,立即检测。
(4)ELISA操作方法:①将RD5-39稀释液200 μL加入NSB(非特异性结合孔,只加样品,不加抗体)。RD5-39稀释液150 μL加入B0孔。将待检测样品、标准品工作液加入96孔酶标板中,每孔150 μL,每个待测样品和标准品设三个复孔;②每孔依次加入50 μL PGE2的一抗(NSB除外),50 μL PGE2Conjugate,粘附贴条覆盖,室温下避光孵育2 h;③小心除去孔内的混合液,每孔加入400 μL稀释后洗脱液,停留数分钟后,将96孔板倒置于干净的纸巾上,多次扣板,到纸巾上无水渍出现为止;共洗4次;④每孔加入200 μL底物反应溶液,室温避光孵育20 min后,每孔加入50 μL反应终止液,微震96孔板,充分混匀;⑤放于酶联免疫检测仪450 nm处测量各孔的吸光值,校正波长为570 nm;⑥根据PGE2标准品所测出的OD值,制作PGE2含量标准曲线,根据计算公式推算待检样品中PGE2的含量。
4.流式细胞仪检测细胞周期
取对数生长期SW1116/HCPT细胞,3×105/孔接种于六孔板,24 h后给予不同浓度的PGE2进行干预,50 μg/mL的PGE2储存液,分别取5 μL、0.5 μL加入2 mL RPMI-1640无血清培养液中,使PGE2的最终浓度分别为10-7mol/L,10-8mol/L。干预48 h后上流式细胞仪检测。每个样品设3个复孔,实验重复3次。
三、统计学处理
结果
一、COX-2在多药耐药SW1116/HCPT细胞中的mRNA水平和蛋白表达水平明显下降,且伴随其活性降低
Realtime PCR重复3次检测COX-2在SW1116细胞,多药耐药SW1116/HCPT细胞中的相对表达,相对比为5.65:1;12.895:1;8.195:1;平均8.9133±3.6755;P<0.05,差异显著。同SW1116细胞相比,在耐药细胞中,COX-2的mRNA水平和蛋白表达水平,均降低;即使增加上样量SW1116/ HCPT中COX-2蛋白表达仍很低(如图1);同时用ELISA检测两种细胞在同样的培养条件下,细胞培养上清中PGE2的微量变化,在SW1116/HCPT耐药细胞中,PGE2含量明显降低([2.5892±0.1510)ng/ mL vs(0.5137±0.0845)ng/mL,P=0.0034];间接反映了COX-2在两种细胞中的活性差别(如图2)。

图1 COX-2的mRNA和蛋白表达差异

图2 SW1116、SW1116/HCPT细胞培养上清中PGE2含量
二、PPARα激动剂WY-14643促进COX-2蛋白表达,提高COX-2活性
为了进一步研究PPARα对COX-2调节作用,我们选择COX-2表达相对比较低SW1116/HCPT细胞,给予PPARα激动剂WY-14643进行干预,用DMSO稀释至终浓度分别为50 μM、100 μM、250 μM,对照组均加同等浓度的DMSO(设为0 μM);见图3F中,WY-14643干预后,同对照组相比,COX-2蛋白在100 μM、250 μM表达明显升高,在50 μM浓度无明显变化;100 μM和250 μM浓度相比,蛋白表达水平无明显变化。见图3A中,50 μM组,PGE2含量为(0.2675±0.03658)ng/mL vs(0.2443±0.01228)ng/mL(对照0 μM);P值为0.2325,无显著差异;图3B中,100 μM:(0.5483± 0.0614)ng/mL,P值分别为0.0083,差异显著;图3C中,250 μM:(0.7851±0.0203)ng/mL,P值为0.000,差异显著。此外,我们还进行了不同浓度之间的组间比较:图3D中,50 μM组同100 μM组相比,P值为0.0025;图3E中,250 μM浓度组同100 μM浓度组相比,P值为0.0098,说明激动剂WY-14643在增加COX-2蛋白表达的同时,也提高了COX-2酶的活性,进而促进PGE2的生成。随着干预浓度的增加,PGE2的合成也逐渐增多,存在剂量依赖相关性。
三、PPARα表达下调抑制COX-2蛋白表达,减少PGE2的合成
在之前的研究中建立了PPARa干扰质粒[4],载体为p-RNAT-U6.1/neo,shRNA 5′CGATCAAGTGACATTGCTAAA 3′。如图4A所示,PPARa干扰质粒下调PPARα蛋白表达,COX-2的蛋白表达也有所下降;图4B中,同SW1116的转染试剂阴性对照组相比,PPARα表达下调后PGE2的含量均明显下降。24 h组PGE2含量由对照组的(2.9405± 0.7167)ng/mL降低为(0.2818±0.0330)ng/mL;48 h组,由对照组的(2.2300±0.2468)ng/mL下降至(0.5533±0.0100)ng/mL,统计P值分别为0.0345和0.0107。
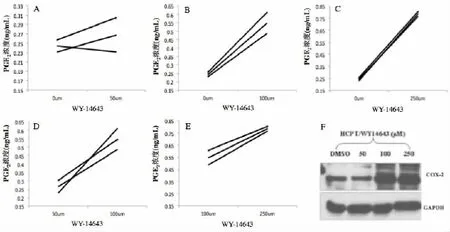
图3 WY-14643对COX-2蛋白表达和酶活性影响的测定

图4 PPARα表达下调对COX-2蛋白表达和活性的影响
四、PGE2促进SW1116/HCPT细胞的增殖
细胞周期的观察指标有细胞增殖指数(Proliferation index,PI)和S期细胞分数(S-phase cell fraction,SPF)。PI反映处于增殖期状态的细胞所占总细胞的百分数。增殖指数PI=(S+G2/M)/(G0/G1+S +G2/M)×100%。如图5所示,给予不同浓度PGE2干预后,G1比例逐渐降低,而S比例逐渐升高,PI由对照组的(32.2±2.13)%,上升到(42.2±3.14)%、(52.1±2.43)%,实验重复3次后,通过对细胞周期PI的多样本均数方差分析,P值为0.0123,差异有统计学意义。结果提示PGE2可能通过对细胞周期的改变来促进细胞增殖。在人结肠癌羟基喜树碱多药耐药SW1116/HCPT细胞株中,由于COX-2表达水平的下调,我们推测在该细胞中,PGE2的合成也应该是减少的,并且通过我们对SW1116细胞和SW1116/HCPT细胞培养上清的ELISA检测,结果显示PGE2的浓度在后者也是非常低的。

图5 PGE2对细胞周期的影响
讨论
过氧化物酶体增殖物激活受体(Peroxisome proliferator-activated receptors,PPARs)参与调控脂质代谢、能量平衡和炎症反应的基因表达。研究已表明,PPARα是肝脏线粒体、过氧化物酶体β-氧化和微粒体ω氧化的主要调节器[5]。在人类,PPARα主要表达于心、肝、肾近曲小管、骨骼肌和棕色脂肪等代谢活跃组织;另外,在单核细胞、血管内皮细胞及血管平滑肌细胞有较低水平表达[6]。
环氧化酶(COX)是前列腺素合成过程中的重要限速酶。COX-2是诱导型同工酶,静息时不表达,在各种细胞因子、癌基因诱导下高表达[7],产生大量COX-2,促进组织大量合成前列腺素(prostaglandins,PGs),尤其是前列腺素E2(prostaglandin E2,PGE2)介导炎症、疼痛、发热等病理生理过程,通过促进细胞增殖、血管形成、抑制细胞凋亡及免疫功能等机制,参与肿瘤发生和发展。在胃癌[8]、肺癌[9]、前列腺癌[10]、胰腺癌[11]等肿瘤组织中高表达。近年来研究显示COX-2不仅与结、直肠癌的发生密切相关,促进肿瘤的转移与侵袭[12-13],COX-2作为一个促癌基因已受到广泛研究和关注。COX-2还受PI3-K/mTOR等信号通路的调节,在脂质代谢方面发挥调节作用[14]。
人结肠癌羟基喜树碱SW1116/HCPT耐药细胞株,是研究结肠癌多药耐药的理想模型[4,15]。运用基因芯片技术对比研究了SW1116/HCPT耐药细胞株与非耐药SW1116细胞株在mRNA水平的差异,发现SW1116/HCPT细胞中存在多种耐药相关基因表达改变,其中PPARα下调倍数高达105倍[3]。研究表明PPARα表达缺失导致对HCPT耐药的抗肿瘤效应,miR-506过表达可以通过抑制PPARα的表达,促进SW1116/HCPT耐药细胞株对HCPT的耐药[4]。我们的研究发现,同SW1116细胞株相比,SW1116/HCPT耐药细胞株中COX-2的mRNA水平下降了8倍,蛋白水平也明显下降,并且COX-2的蛋白表达随着PPARα表达下调而下降。为了进一步研究PPARα对COX-2的调节作用,我们采用更敏感的ELISA检测法,检测细胞培养上清中PGE2的含量,来间接反应COX-2的活性。结果发现PPARα激动剂WY-14643在促进COX-2蛋白表达的同时也提高了COX-2的酶活性表达,表现为PGE2含量的剂量依赖性升高;我们还利用RNA干扰技术,下调PPARα的表达后,再次检测PGE2的含量,结果发现,同SW1116转染阴性对照质粒组相比,转染PPARα干扰质粒的SW1116细胞,培养上清中PGE2的含量由2.94 ng/mL降低到0.28 ng/ mL;从这两方面来看,PPARα对COX-2可能是存在着正向调节的关系,即PPARα可以促进COX-2的表达,PPARα的下调同样也抑制了COX-2的表达。因此我们推测在结肠癌多药耐药SW1116/ HCPT耐药细胞中,存在PPARα/COX-2/PGE2的调节机制,COX-2很可能是PPARα的靶基因。
有关COX-2的研究已经发现,阿司匹林及其他非甾体类抗炎药(non-steroidal anti-inflammatory drugs,NSAIDs)能降低大肠癌的发病率及死亡率并且与COX-2的活性受到抑制有关[16]。而COX-2受到抑制后,其产物PGE2的合成必将减少。目前的研究发现PGE2可以通过抑制TNF-α、IFN、IL-1等促进肿瘤生长,诱导血管生成,并且可以与细胞膜受体结合或者直接进入细胞核,与细胞核受体如PPARδ结合,诱导K-ras、H-ras、Bcl-2等促癌基因的表达[17]。本研究的结果也证明,PGE2可以促进结肠癌细胞的增殖,因此COX-2的选择性抑制剂对结肠癌细胞的生长和侵袭有一定的抑制作用,COX-2选择性抑制剂具有应用于临床治疗结肠癌的潜在价值。已有研究表明,COX-2抑制剂双氯芬酸,可以抑制肿瘤中PI3-K,AKT的表达,下调抗凋亡蛋白BcL-2的表达,上调促凋亡蛋白Bax、caspase-3 and cas-pase-9的表达,从而促进肿瘤细胞的凋亡[18]。
在我们的人结肠癌羟基喜树碱多药耐药细胞株SW1116/HCPT细胞中,PPARα的表达明显低于亲代SW1116细胞,如果从PPARα对COX-2正向调控来说,COX-2表达的下调似乎可以用PPARα表达的下调来解释。国外也有相关的类似研究[19]发现过氧化物酶体(PP)可以促进COX-2的表达,这与我们的实验发现基本上是一致的。此外,国外亦有文献报道脱氧胆酸盐可以通过ERK1/2-P38-MAPKAP-1信号通路,诱导COX-2的表达[20]。有研究表明,PPARα激动剂非诺贝特,可以通过抑制氧化应激、p38MAPK和JNK的磷酸化,减少自发性高血压大鼠的肾脏损害[21]。PPARα激动剂还可以通过抑制VEGF诱导的AKT的磷酸化,发挥抑制内皮细胞迁移的能力[22]。亦有研究表明,PPARα激动剂WY-14643可以促进ERK、P38的磷酸化,而ERK信号通路抑制剂PD98059和U0126则能完全抑制WY-14643诱导的纤维蛋白溶酶原激活剂抑制剂(plasminogen activator inhibitor 1,PAI-1)的释放[23]。根据以上的理论,WY-14643诱导COX-2表达增加的机制则可能是通过ERK1/2-P38-MAPK-AP-1-COX-2信号通路来实现的[20]。
1 Tsujii M,Kawano S,Tsuji S,et al.Cyclooxygenase regulates angiogenesis induced by colon cancer cells.Cell,1998,93(5):705-716.
2 Fukuda R,Kelly B,Semenza GL.Vascular endothelial growth factor gene expresion in colon cancer cels exposed to prosta-glandin E2is mediated by hypoxia-inducible factor 1.Cancer Res,2003,63(9):2330-2334.
3 邹健,冉志华,黄美兰,等.人结肠癌羟基喜树碱耐药细胞株SW1116/HCPT的建立与鉴定.胃肠病学,2006,11(6):327-331.
4 Tong JL,Zhang CP,Nie F,et al.MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells.FEBS Lett,2011,585(22):3560-3568.
5 Souza-Mello V.Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease.World J Hepatol,2015,7(8):1012-1019.
6 Forman BM,Chen J,Evans RM,et al.The peroxisome proliferatoractivated receptors:ligands and activators.Ann N Y Acad Sci,1996,804:266-275.
7 Zhan J,Liu JP,Zhu ZH,et al.Relationship between COX-2 expression and clinicopathological features of colorectal cancers.Chin Med J,2004,117(8):1151-1154.
8 Ristimäki A,Honkanen N,Jänkälä H,et al.Expression of cyclooxygenase-2 in human gastric carcinoma.Cancer Res,1997,57(7):1276-1280.
9 Huang M,Stolina M,Sharma S,et al.Non2 small cell lung cancer cyclooxygenase-2 dependent regulation of cytokine balance in lymphocytes and macrophages:up-regulation of interleukin 10 and down-regulation of interleukin 12 production.Cancer Res,1998,58(6):1208-1216.
10 Tjandrawinata RR,Dahiya R,Hughes-Fulford M.Induction of cyclooxygenase-2 mRNA by prostaglandin E2 in human prostatic carcinoma cells.Br J Cancer,1997,75(8):1111-1118.
11 Tucker ON,Dannenberg AJ,Yang EK,et al.Cyclooxygenase-2 expression is upregulated in human pancreatic cancer.Cancer Res,1999,59(5):987-990.
12 Zhang H,Sun XF.Overexpression of cyclooxygenase-2 correlates with advanced stages of colorectal cancer.Am J Gastroenterol,2002,97(4):1037-1041.
13 Eberhart CE,Coffey RJ,Radhika A,et al.Up-regulation of cyclooxygenase-2 gene expression in human colorectal adenomas and adenocarcinomas.Gast roenterology,1994,107(4):1183-1188.
14 Fazolini NP,Cruz AL,Werneck MB,et al.Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation,cytokine production and increased cell proliferation.Cell Cycle,2015,14(16):2667-2676.
15 Zhu MM1,Tong JL,Xu Q,et al.Increased JNK1 signaling pathway is responsible for ABCG2-mediated multidrug resistance in human colon cancer.PLoS One,2012,7(8):e41763.
16 Thun MJ,Namboodiri MM,Calle EE,et a1.Aspirin use and risk of fatal cancer.Cancer Res,1993,53(6):1322-1327.
17 Prescott SM,Fitzpatrick FA.Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta,2000,1470(2):M69-78.
18 Rana C,Piplani H,Vaish V,et al.Downregulation of PI3-K/Akt/ PTEN pathway and activation of mitochondrial intrinsic apoptosis by Diclofenac and Curcumin in colon cancer.Mol Cell Biochem,2015,402(1-2):225-241.
19 Ikawa H,Kameda H,Kamitani H,et al.Effect of PPAR activators on cytokine-stimulated cyclooxygenase-2 expression in human colorectal carcinoma cells.Exp Cell Res,2001,267(1):73-80.
20 Looby E,Abdel-Latif MM,Athié-Morales V,et al.Deoxycholate induces COX-2 expression via Erk1/2-,p38-MAPK and AP-1-dependent mechanisms in esophageal cancer cells.BMC Cancer,2009,9(6):190.
21 Hou X,Shen YH,Li C,et al.PPARalpha agonist fenofibrate protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress and MAPK activity.Biochem Biophys Res Commun,2010,394(3):653-659.
22 Goetze S,Eilers F,Bungenstock A,et al.PPAR activators inhibit endothelial cell migration by targeting Akt.Biochem Biophys Res Commun,2002,293(5):1431-1437.
23 Banfi C,Auwerx J,Poma F,et al.Induction of plasminogen activator inhibitor I by the PPARalpha ligand,Wy-14,643,is dependent on ERK1/2 signaling pathway.Thromb Haemost,2003,90(4):611-619.
(本文编辑:郭彦东)
Role of PPARα/COX-2/PGE2pathway in human drug resistant colon cancer cell line
NIE Fang,ZHAO Xian-yuan,YU Yue-tian,YIN Rong,LI Yu-jie,CAO Jian-guo,GAO Yuan.Department of Intensive Care Medicine,Ren Ji Hospital,School of Medicine,Shanghai Jiao Tong University.Cao Jian-guo:145 Shandong Middle Rd.Shanghai 200001.E-mail:doctorcjg@hotmail.com.Gao Yuan:1630 Dongfang Rd.Shanghai,China.E-mail:gaoyuanzhuren@126.com.
10.3969/j.issn.1672-2159.2015.06.006
200001上海交通大学医学院附属仁济医院重症医学科
曹建国,E-mail:doctorcjg@hotmail.com;皋源,E-mail:gaoyuanzhuren@126.com
上海高校青年教师培养资助计划(shjdy043)
2015-07-23)
