抗肿瘤候选药物SPH0396对CYP1A2和CYP3A4的诱导研究
2015-09-14徐佳向志雄万惠新夏广新上海医药集团股份有限公司中央研究院上海201203
徐佳向志雄 万惠新 夏广新(上海医药集团股份有限公司中央研究院 上海 201203)
抗肿瘤候选药物SPH0396对CYP1A2和CYP3A4的诱导研究
徐佳*向志雄 万惠新 夏广新**
(上海医药集团股份有限公司中央研究院 上海 201203)
目的:建立稳定高效的原代大鼠肝细胞和冻存人肝细胞诱导实验模型,研究SPH0396对CYP1A2和CYP3A4的诱导。方法:利用探针底物代谢物生成量对CYP3A4和CYP1A2的酶活性进行评价。结果:SPH0396在0.1mol/L和1mol/L对CYP酶(1A2和3A4)无诱导作用,3mol/L时对CYP3A4无诱导,而对CYP1A2的诱导倍数均大于2,初步认为其诱导CYP1A2,且诱导作用在原代大鼠与冻存人肝细胞中无种属差异。结论:SPH0396能诱导CYP1A2,需关注SPH0396因诱导CYP1A2而导致临床上发生药物-药物相互作用的可能性。
酪氨酸激酶抑制剂 肝细胞 诱导
酪氨酸激酶(tyrosine kinase,TKs)在许多生理过程中都发挥重要作用,而TKs的活性失调被证明与多种癌症的发生与发展相关。近年来,以酪氨酸激酶为靶点进行药物研发已成为国际上抗肿瘤药物研究的热点,多种小分子酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)被批准上市用于肿瘤治疗[1-2]。针对Bcr-Abl靶点,我院设计并合成了一系列化合物,并对其中部分化合物进行了体外药效学和早期代谢性质的评价。其中SPH0396(图1)在激酶和细胞水平的抗肿瘤活性均接近或优于上市药物bosutinib[3-5],体内外药代性质良好,因此将其选为候选化合物进一步研究。
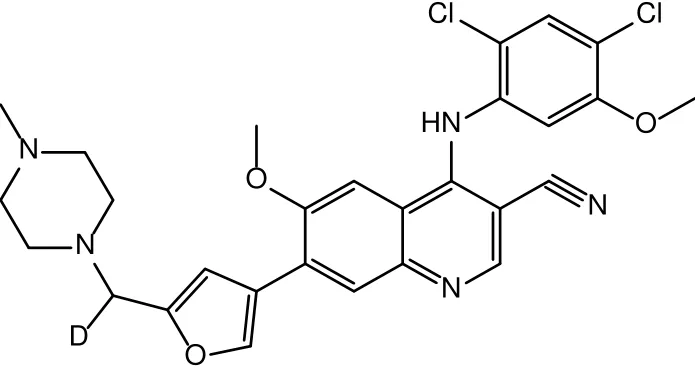
图1 SPH0396的化学结构
药物间的相互作用(drug-drug interactions,DDI)指的是某种药物的作用时间或者作用强度在与其他药物同时服用时发生了药效学和药代学上可以量化评价的变化。药代动力学方面的DDI很难被发现:绝大多数的药物通过肠道和/或肝细胞色素P450酶代谢,在被代谢酶抑制或诱导或转运的的过程中,可能发生DDI[6]。近20年来,美国FDA已先后将批准上市的数十种新药从市场撤出,其主要的原因就是出现了严重的药物代谢性相互作用。诱导CYP450酶的表达或功能上调,加快药物的代谢速率影响临床疗效或引起中毒。诱导的主要作用机制是通过核受体介导的基因转录水平的提高,其中芳香烃受体(AhR)介导CYP1A2,孕烷X受体(PXR)介导CYP3A、CYP2C、CYP2B,组成型雄甾烷受体(CAR)介导CYP2B6[6-8]。因CYP2B6诱导剂苯巴比妥为管制药品,且CYP3A、CYP2C、CYP2B为同一受体介导,新药早期诱导作用的评价选择CYP1A2和CYP3A4。
早期诱导作用评价可以避免在漫长的药物研发过程中因为诱导造成大量问题没有得到及时发现和解决而造成的损失。体外肝细胞培养作为观察药物是否对肝微粒体酶亚型有诱导作用的体外实验模型,用来预测DDI,具有可较经济和迅速地阐明药物对药物代谢酶的诱导,从而避免采用大量动物进行药理效应筛选,同时易排除体内多种因素的影响及便于作专项研究等优点。新药对CYP450酶的诱导作用已越来越受到国际医药界的重视,成为FDA对CYP450酶诱导能力评价的标准之一[8]。
1 材料与方法
1.1 材料
1.1.1 动物
SD大鼠一只(上海西普尔必凯实验动物有限公司,许可证号SCXK(沪)2013~0016),雄性,体重200 g。
1.1.2 药品与试剂
William’s E Medium、谷氨酰胺(292 mg/L)、双抗溶液(含青霉素10 000 U/ml,链霉素10 000 mg/ml)(5 ml/500 ml)、胰岛素(8 mg/ml)、5% 胎牛血清(0.22 mm微孔滤膜过滤除菌,4 ℃保存);PBS预灌流溶液(Dulbecco's phosphate buffered saline(10x))稀释10倍,EDTA 0.1 mmol/L,NaHCO3调pH 7.2~7.4,0.22 mm微孔滤膜过滤除菌,4 ℃保存;鼠尾胶原(0.05 mg/ml)用0.02 mol/L的醋酸稀释,临用现配;以上试剂均购自Gibco公司; Overlay(0.25 mg/mL的Matrigel(BD)(用William’s E 培养基稀释,临用现配);地塞米松与利福平购自中国药品生物制品检定所,0.05%胶原酶Ⅳ溶液(D-Hanks溶液溶解,0.22 mm微孔滤膜过滤除菌)、Krebs-Henseleit buffer Modified、β-萘黄酮、奥美拉唑、非那西汀、睾酮均购自美国Sigma公司,试验用水为灭菌去离子水,其他试剂均为国产分析纯及以上级别。
1.1.3 实验仪器
48孔培养板、CellBIND Surface Products 48孔表面培养板购自Corning公司;生物安全柜(Labconco公司);CO2恒温培养箱(Thermo公司);TS100型倒置显微镜(日本尼康公司);Sigma 3K15离心机;CBS-47液氮罐;ZHWY-103B摇床(上海智城分析仪器制造有限公司);Thermo Varioskan Flash 酶标仪,API4000 QTRAP型串联质谱仪,配有电喷雾离子源(ESI)以及Analyst 1.5.1数据处理软件(美国Applied Biosystem公司);Waters Acquity UPLC系统,包括四元输液泵和自动进样器(美国Waters公司)。
1.1.4 溶液配制
D-Hanks溶液(g/L):CaCl20.55,NaCl 8.0,KCl 0.4,KH2PO40.06,Na2HPO4·7H2O 0.06,NaHCO30.35,超纯水溶解,NaHCO3调pH 7.2~7.4,4 ℃保存。
0.02 mol/L的醋酸:115 ml的醋酸(大于99%)用水稀释至100 ml,0.22 mm微孔滤膜过滤除菌。
Krebs-Henseleit buffer(KHB):Krebs-Henseleit buffer Modified (Sigma) 9.6 g/L,NaHCO32.2 g/L,用NaHCO3调pH至7.3。
诱导液(阳性化合物)用William’s E 培养基稀释至地塞米松(10mol/L)、利福平(20mol/L)、β-萘黄酮(50mol/L)、奥美拉唑(100mol/L)。
探针底物非那西汀(CYP1A2)和睾酮(CYP3A4)用KHB稀释到100mol/L和200mol/L。
1.2 方法
1.2.1 大鼠原代肝细胞与冻存人肝细胞诱导实验模型的建立
1.2.1.1 实验准备
方法1 在48孔板底涂布鼠尾胶原(0.05 mg/ml)300 ml/孔,置于CO2恒温培养箱成胶后吸出多余胶原(时间视具体情况而定),细胞悬液种板前用William’ s E培养基洗去表面的酸性物质。
方法2 CellBIND Surface Products 48孔表面培养板直接种细胞。
1.2.1.2 大鼠原代肝细胞诱导实验
以原位二步IV型胶原灌注法消化大鼠的肝脏[9-10],低速离心法分离肝实质细胞
腹腔注射水合氯醛麻醉大鼠(0.5 ml/100 g),仰卧固定,门静脉插入导管,动脉夹固定,经门静脉插管以0.5 ml/min的速度匀速灌注预温孵的PBS预灌流溶液以去除肝内血液及钙离子,当肝脏变苍白、下腔静脉流出液体变清时开始灌注预温的胶原酶液,流速约0.1 ml/min,约6~10 min,肝脏颜色变成土黄色、包膜下呈龟背状裂隙、压之凹陷不易恢复时停止灌注,完整摘除肝脏。超净台中揭去被膜,将细胞混悬液用200目尼龙网筛过滤,滤液50 g低速离心3 min,沉淀用William’s E 培养基离心1次(50 g,3 min),接着同法洗涤2次,最后加入William’s E 培养基制成细胞悬液,预先在每孔中加入100 ml Williams’E 培养基,以防止细胞聚集,按1×106个/ml的细胞密度分别种板(CellBIND48孔表面培养板与包被鼠尾胶原的48孔表面培养板),每孔100 ml,2~6 h后(视细胞贴壁状态而定)换William’s E培养基,间隔24 h换William’s E 培养基(含0.25 mg/ml的Matrigel)500 ml/孔,以后每天换1次William’s E 培养基(500 ml/孔),至第3天。第4天加入诱导剂(诱导剂用William’s E培养基稀释,0.22 mm微孔滤膜过滤除菌),每天换液(诱导剂)至第6天。
1.2.1.3 冻存人肝细胞诱导实验
细胞复苏,低速离心(50×g,3 min),洗涤2次(William’s E 培养基),最后加入William’s E 培养基制成细胞悬液,细胞贴壁后换William’s E 培养基(含0.25 mg/ml的Matrigel)500 ml/孔,第2天加入诱导剂(每天换液(诱导剂)至第5天)。
1.2.1.4 诱导实验及样品处理方法
加药前各孔加入37 ℃的空白KHB 1 ml,置于37 ℃摇床中,10 min后轻轻吸干孔内KHB,以期在不破坏其单层膜的基础上除去细胞表面的杂质。将底物按设定浓度溶解于KHB液中,后加入各孔(48孔板加入100 ml/孔),置于37 ℃培养箱中,60 min时迅速吸出药物,同时做空白对照(加入空白KHB)。取出一定量的样品,加入等体积的含替硝唑(0.1 mg/ml)的乙腈,混合10 min沉淀蛋白,离心(6 000 g,10 min)一次,取50 ml进板。
1.2.2 样品测定
利用探针底物的代谢物(睾酮代谢物为6-羟基睾酮,非那西汀代谢物为对乙酰氨基酚)生成量对CYP3A4和CYP1A2的酶活性进行评价。
色谱柱为Xterra C18柱(2.1 mm×50 mm,1.7 mm,美国Waters公司)。流动相A为0.1%甲酸-水、B为0.1%甲酸-乙腈,洗脱程序及流动相A-B配比如下:0~0.5 min 95∶5,0.5~1.0 min线性变至5∶95,1.0~2.0 min维持5∶95,2.0~2.5 min线性恢复至95∶5,2.5~3 min保持95∶5。流速0.3 ml/min,柱温30 ℃,进样5 ml。
离子源:电喷雾离子源(ESI),检测方式:正离子检测;离子源温度(TEM):400 ℃;雾化气(Gas1,N2)压力:50 psi;辅助气(Gas 2,N2)压力:50 psi;气帘气(CUR)压力:10 psi;扫描方式为多重反应监测(MRM);碰撞气(CAD,N2)压力:Medium;用于定量分析的离子反应如表1。
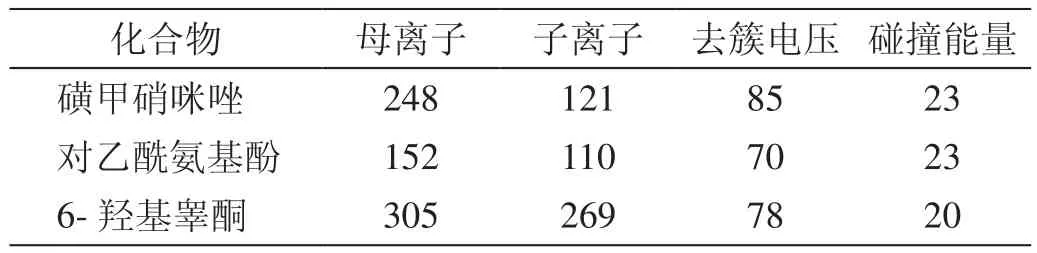
表1 质谱条件
1.2.3 数据处理[8]
相对酶活性 (% of NC)= 测试物给药组或阳性对照组酶活性/阴性对照组酶活性× 100%。
公式中:相对酶活性反映化合物诱导CYP1A2和CYP3A4的能力,指加入化合物诱导与空白对照同时加入探针底物后代谢物生成量之比。
% 相对阳性对照活性=(给测试物组样本活性-阴性对照组样本活性)/(阳性对照组样本活性-阴性对照组样本活性)× 100%。
公式中:化合物与阳性药作为诱导剂同时扣除空白对照,加入相同探针底物后代谢物生成量之比。
FDA规定阳性化合物的相对酶活性要大于2,同时规定化合物的相对酶活性大于等于阳性化合物的40%即为对CYP酶有诱导作用。
2 实验结果
2.1 光学显微镜观察结果
大鼠肝细胞在CELLBIND48孔表面培养板(图2A)与包被鼠尾胶原的48孔表面培养板(图2B)种板后培养24 h后显微镜下观察,从细胞形态分析,两者差异不明显。

图2 显微镜下大鼠肝细胞形态(40×)
2.2 诱导实验结果
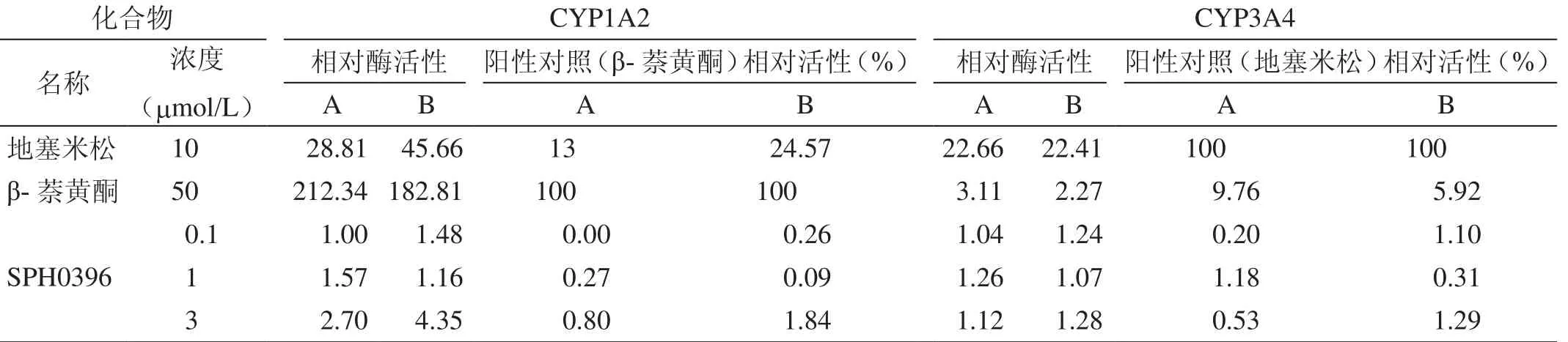
表2 原代大鼠肝细胞诱导实验
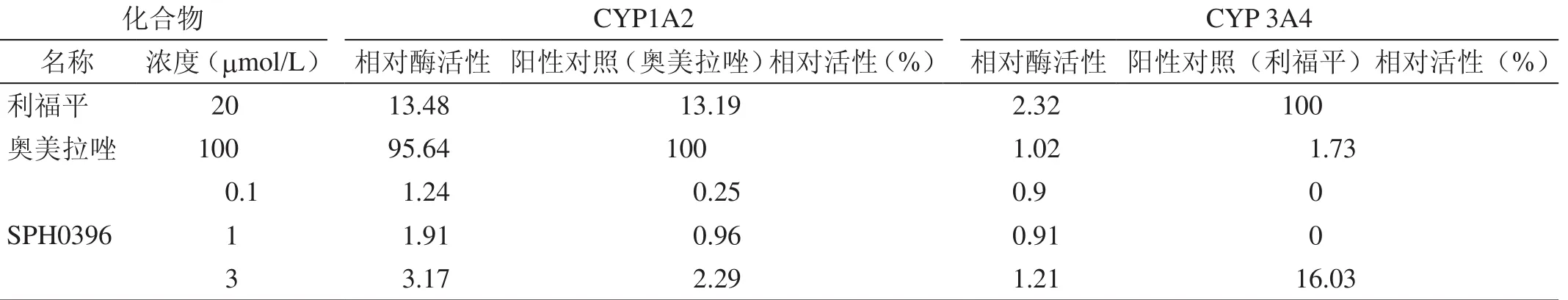
表3 冻存人肝细胞诱导实验结果(CELLBIND板)
3 讨论
肝细胞培养作为一种体外模型,有其突出的优点:与体内情况保持一致,可以在接近生理状态的情况下研究药物的代谢、毒性、预测药物-药物相互作用的可能,排除许多复杂因素的干扰,基本保留肝脏原有的代谢功能和细胞分化状态,具有与体内一致的细胞色素P450酶活性。但是模型的建立过程复杂,经过探索,诱导实验主要需要注意以下几点:①肝细胞接种后2 h开始贴壁,细胞过多会产生接触抑制,贴壁率降低,而细胞培养板或多或少都有中间聚集效应,同时由于肝细胞是实质细胞,不易均匀分散种板,一般推荐采用“星型”摇板技术以均匀分散细胞,但是频繁摇板不利于细胞贴壁[10-11]。本实验采用预先加入100 ml William’s E 培养基后再加入悬浮细胞,可防止细胞聚集,效果良好;②细胞外基质对肝细胞的生长状态有重要的影响:胶原对细胞的贴壁生长很有帮助,但自行包被胶原,价格高、耗时长且稳定性差。购买包被好胶原的培养板价格昂贵,保质期短且需冷藏保存。CellBIND是Corning公司的专利技术,利用强微波等离子体处理培养表面,可以使培养表面结合更多的氧基,从而增强表面的亲水性和稳定性,改善细胞的贴壁,提高细胞产量。相对于用生化包被的方法,CellBIND表面具有更高的稳定性,而且不需冷藏或其它特殊处理。实验比较了细胞在CellBIND Surface Products 48孔表面培养板与包被鼠尾胶原的48孔表面培养板培养的结果,从细胞形态和诱导实验结果分析,两者差异不明显,故采用CellBIND板以便简化实验过程。
FDA规定化合物的相对酶活性≥阳性化合物的40%即为对CYP酶有诱导作用,同时规定阳性化合物的相对酶活性要大于2[8],SPH0396在0.1mol/L和1mol/L的浓度范围内对主要的CYP酶(1A2和3A4)无诱导作用,3mol/L时对CYP 3A4无诱导作用,对CYP1A2的相对酶活性均大于2,分别为2.7、4.35、3.17,尽管不到阳性化合物的40%,但我们认为其诱导CYP1A2,且原代大鼠与冻存人肝细胞无种属差异,所以需要关注因诱导CYP1A2而导致临床上发生药物-药物相互作用的可能性。
[1] Levitzki A. Tyrosine kinase inhibitor: views of selectivity sensitivity, and clinical performance[J]. Annu Rev Pharmacol Toxicol, 2013, 53: 161-185.
[2] 李欣, 高金恒,陈国良. 蛋白酪氨酸激酶抑制剂的研究进展[J]. 沈阳药科大学学报, 2011, 28(12): 1005-1021.
[3] 刘舒畅, 张健存, 陈赛娟. BCR-ABL蛋白酪氨酸激酶抑制剂的研究进展[J]. 中国医药工业杂志, 2010, 41(4): 293-297.
[4] 郑敏, 白秋江, 胡跃明. 慢性髓性白血病治疗药博舒替尼[J]. 药物流行病学杂志, 2014, 23(1): 58-60.
[5] Rabbani SA, Valentino ML, Arakelian A, et al. SKI606 (Bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis[J]. Mol Cancer Ther, 2010, 9(5): 1147-1157.
[6] 郑友广, 李铭东, 吉民. 酪氨酸激酶及其小分子抑制剂研究进展[J]. 药品评价, 2006, 3(2): 137-140.
[7] 高志伟, 施孝金, 钟明康. 细胞色素P450酶与药物相互作用研究进展[J]. 中国临床药学杂志, 2006, 15(6): 395-398.
[8] Guidance for industry: drug interaction studies — study design, data analysis, implications for dosing, and labeling recommendations[EB/OL]. [2015-04-04]. http://www.fda. gov/downloads/drugs/guidancecomplianceregulatoryinformat ion/guidances/ucm292362.pdf.
[9] Seglen PO. Preparation of isolated rat live cells [J]. Methods Cell Biol, 1976, 13: 29-83.
[10] Kono Y, Yang S, Roberts EA. Extended primary culture of human hepatocytes in collagen gel sandwich system [J]. In Vitro Cell Dev Biol Anim, 1997, 33(6): 467-472.
[11] Gómez-Lechón MJ, Lahoz A, Gombau L, et al. In vitro evaluation of potential hepatotoxicity induced by drugs[J]. Curr Pharm Des, 2010, 16(17): 1963-1977.
Study on the induction of anti-tumor candidate SPH0396 to CYP1A2 and CYP3A4
XU Jia*, XIANG Zhixiong, WAN Huixin, XIA Guangxin**
(Central Research Institute, Shanghai Pharmaceuticals Holding Co. Ltd., Shanghai 201203, China)
Objective: To establish a stable and efficient experimental model using primary rat hepatocyte and cyropreserved human hepatocyte so as to study the induction of small molecule compound SPH0396 to CYP1A2 and CYP3A4. Methods: The enzyme activity was evaluated using probe substrates metabolite. Results: SPH0396 had no effect on CYP3A4 at the concentrations of 0.1 ~ 3mol/L, suggesting that SPH0396 is not an inducer of CYP3A4. However, significant induction of CYP1A2 was observed at 3mol/L of SPH0396 and the induction reached more than two times. There is no difference between rat and human hepatocytes. Conclusion: SPH0396 may cause drug-drug interaction via induction of CYP1A2 and attention should be paid in clinic to the potential of drug-drug interaction resulting from its induction to CYP1A2.
tyrosine kinase inhibitors; hepatocyte; induction
R969.2; R979.19
A
1006-1533(2015)19-0071-05
徐佳(1984-),女,工程师,主要从事药代动力学方面的新药研发工作。E-mail:xujia@ sphchina.com
**通讯作者:夏广新,男,教授级高级工程师,主要从事新药研发工作。E-mail:xiagx@sphchina.com
2015-06-03)
