硫酸氢氯吡格雷球形结晶工艺及其生长机理
2015-08-22王海洋杜艳妮李振方宋晓鹏谭端明龚俊波
王海洋,杜艳妮,李振方,宋晓鹏,谭端明,龚俊波
(1天津大学化工学院化学工程联合国家重点实验室,天津 300072;2天津化学化工协同创新中心,天津 300072; 3深圳信立泰药业股份有限公司,深圳 518102)
引 言
多晶型是有机药物中广泛存在的一种现象。随着分析技术的发展,越来越多的药物被发现存在多晶型现象[1]。对于多晶型药物来说,同一药物不同晶型的产品可能存在晶体内部分子间作用力以及表面性质的差异,其理化性质如溶解度、熔点、密度、硬度、比热容、晶体形态等可能有所差异,这不仅会影响医药产品的流动性、可压缩性、凝聚性能等加工性能,更重要的是还可能引起药物溶出速率、溶出度、稳定性等的质量差异,从而影响药物的生物活性与生物利用度,导致临床疗效的差异[2]。此外,不同晶型受专利保护的情况也一般不同。因此,工业上在进行多晶型药物的制备时,需要注意晶型转化问题,生产特定晶型的纯品。但不同药物体系中转晶所需的条件各异,有些药物只有在溶液中才会发生转晶,药物不同晶型之间溶解度的差异是其转晶的推动力[3-5];而另一些药物则可能发生固态转晶行为,如邻氨基苯甲酸和盐酸维法拉新等[6-8]。此外,添加剂和模板剂的存在也会对转晶过程造成影响[9-12]。药物开发中往往需要得到稳定晶型,以避免在后续操作过程中发生晶型转变的危险;但有时候为了获得高的溶解速率和生物利用度,也需要制备介稳晶型,因此需要研究如何阻止或者延缓介稳晶型向稳定晶型的转变。在这一研究中,过程分析技术应用广泛,如过程在线红外光谱[13]、过程在线拉曼光谱[14]和差示扫描量热仪[15]的应用,使得人们对于转晶过程的理解和控制有了很大进步。
“球形结晶”是由日本的Kawashima等[16]于1982年首次提出的一种技术,在水杨酸的结晶过程中完成了整粒过程,得到的水杨酸球形晶体流动性和堆密度有了很大提高。随后,这种技术在越来越多的药物体系中得到了成功应用。目前,常用的方法有球形聚结[17-18]、准乳液扩散[19-20]、氨扩散[21-22]、晶体共聚[23]和中和滴定[24]等。鉴于球形晶体在流动性、稳定性等方面的巨大优势,球形晶体的制备手段成为了一个研究热点。
硫酸氢氯吡格雷(CHS)是一种常用的血小板抑制剂,只有Ⅰ、Ⅱ晶型有入药价值,且Ⅱ晶型仍受专利保护。在制备硫酸氢氯吡格雷Ⅰ晶型产品时主要存在两个问题:一是晶型不稳定,得到的产品晶型纯度达不到要求;二是产品的堆密度低,流动性差,不利于后续的储存、压片等操作。本研究通过结晶过程分析与优化,成功制备了纯Ⅰ晶型的硫酸氢氯吡格雷球形产品,大大提高了产品的堆密度和流动性,并分析了其球形结晶机理。
1 实验材料和方法
1.1 材料
氯吡格雷樟脑磺酸盐,由深圳信立泰药业有限公司提供,纯度≥99.0%;仲丁醇、乙酸乙酯、二氯甲烷、浓硫酸、无水硫酸镁和碳酸氢钠由天津市江天化工技术有限公司提供,分析纯;Ⅰ晶型的硫酸氢氯吡格雷是在以甲基异丁基酮为溶剂的反应结晶过程中得到,Ⅱ晶型硫酸氢氯吡格雷是用丙酮作为溶剂进行反应结晶得到,X射线粉末衍射均未检测出其他晶型特征峰。
1.2 分析测试仪器
X射线粉末衍射仪,D/Max2500,日本理学株式会社;扫描电子显微镜,TM3000,日本日立公司;偏光显微镜,BX51,日本奥林巴斯公司;在线红外光谱(ATR-FTIR),瑞士梅特勒-托利多公司;电子天平(精度0.1 mg),AL204-IC,瑞士梅特勒-托利多公司;粒度分布仪,Mastersizer3000,英国马尔文公司;恒温水浴,CH1015,上海舜宇恒平仪器有限公司。
1.3 溶解度测量方法
本文使用动态法测定Ⅰ、Ⅱ晶型硫酸氢氯吡格雷在仲丁醇中的溶解度。在1 ml的小瓶中,加入一定量的固体溶质和溶剂,通过磁子搅拌使两相相互混合,随着温度的升高,悬浮液中溶质的溶解度会逐渐增大而最终完全溶解,而溶液的透光率也会由于溶质完全溶解而发生突变,实验中通过对透光率的观察来确定平衡点。设定升温速率(0.25℃·min-1),随着温度的升高,溶质逐渐溶解,当透光率达到100%时记录此时的温度,即为溶解温度。每种晶型测定30个点。根据溶剂与溶质的质量比求得此温度下的溶解度。
1.4 球形结晶过程
具体球形结晶过程如下:(1)称取一定量氯吡格雷樟脑磺酸盐溶于二氯甲烷中,加入饱和碳酸氢钠溶液调节溶液pH为碱性。(2)分液,洗涤有机相,分离得到下层有机相,用无水硫酸镁干燥除水。(3)过滤,旋蒸滤液至恒重,得到氯吡格雷碱。(4)将得到的氯吡格雷碱溶于仲丁醇中,形成一定浓度的游离碱溶液,滴加与游离碱一定摩尔比(硫酸∶游离碱)的浓硫酸进行成盐反应,恒温下加入硫酸氢氯吡格雷Ⅰ晶型晶种,程序控温至实验结束。(5)过滤,乙酸乙酯洗涤滤饼,50℃干燥2 h得到产品。
本文详细考察了结晶温度以及溶剂残留的影响。
2 实验结果与讨论
2.1 硫酸氢氯吡格雷的溶解度
本文采用动态法测量硫酸氢氯吡格雷Ⅰ、Ⅱ两种晶型在仲丁醇中的溶解度,一方面是由于Ⅰ晶型容易发生转晶;另一方面是由于溶解在溶剂中的溶质,在干燥时难以以晶体形态析出,其中会包藏大量溶剂,对称量结果有很大的影响。因此静态法的实验结果重复性不好。采用动态法测定的硫酸氢氯吡格雷溶解度如图1所示。
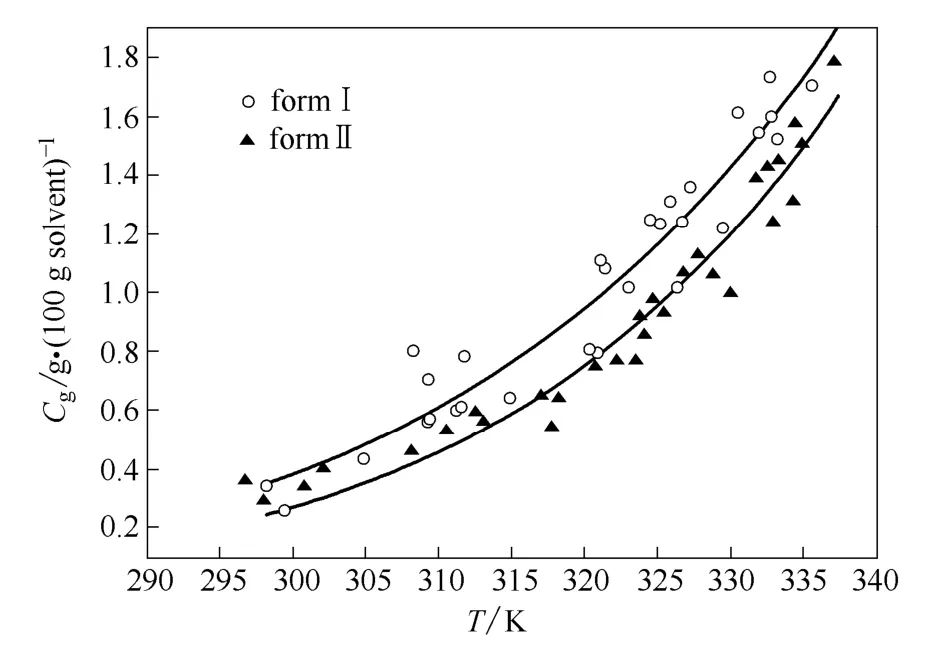
图1 硫酸氢氯吡格雷(CHS)在仲丁醇中的溶解度 Fig.1 Solubility of CHS in 2-butanol
从两种晶型的溶解度中可以看出:(1)两种晶型在仲丁醇中的溶解度都很低,并且在测量范围内,Ⅱ晶型始终为稳定晶型,此体系为单变体系;(2)在各个温度时,Ⅰ、Ⅱ晶型的溶解度差距变化不大,因此转晶的推动力基本为定值。但由于随着温度的提高,晶体的溶解速率与结晶速率均增快,因此在高温下,容易发生转晶,不利于Ⅰ晶型的制备。
2.2 过程优化
根据硫酸氢氯吡格雷在仲丁醇中的溶解度实验确定,在实验的温度范围内,Ⅰ晶型都为介稳晶型,且反应温度不能过高以防止转晶过程过快。本文详细考察了结晶温度和溶剂残留对晶型纯度的影响。
2.2.1 温度 在实验溶剂选定的情况下,温度是影响晶型的关键因素。这是由于在不同温度下,反应速率、晶体生长速率和溶解速率等都会有很大差异,容易对转晶速率产生影响。
在实验中分别考察了25、30和35℃下反应结晶得到的产品,并利用XRD对3组产品的晶型进行表征。表征结果如图2所示。
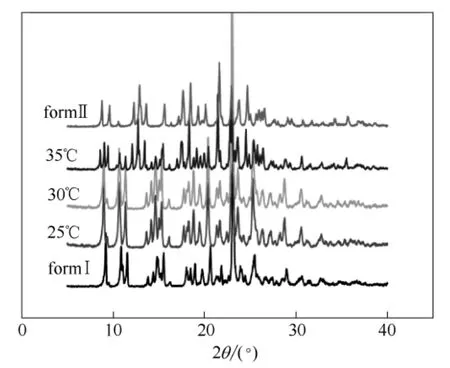
图2 硫酸氢氯吡格雷各温度条件下产品XRD衍射图谱 Fig.2 X-ray powder diffraction patterns of CHS obtained under different temperatures
XRD结果显示,当温度为35℃时,其他实验条件相同的情况下,得到的产品为混晶。可得出,当结晶温度为35℃,达到目标的收率时,产品中已经含有了Ⅱ晶型。因此在制备Ⅰ晶型产品时,应将温度设定在35℃以下。
2.2.2 残留溶剂 在第一步的碱化反应中,需要先将氯吡格雷游离碱溶于水和二氯甲烷中,然后将溶剂除去,分离得到氯吡格雷游离碱。因此,得到的游离碱中可能会含有的溶剂水和二氯甲烷,因此在实验过程中,需要考虑这两种溶剂对晶型的影响。
首先,确定晶型的含量与其XRD特征峰强度之间的关系。从两种晶型的XRD图谱(图3)中可以看出,不同晶型对应不同的特征峰,Ⅰ晶型在2θ=9.1°处有一个特征峰,Ⅱ晶型在12.1°、12.9°和13.5°(2θ)3处的特征峰最为敏感。利用特征峰的强度定量表征Ⅱ晶型在产品中的含量,从而确定杂质溶剂对于晶型的影响。文中选用Ⅰ晶型在9.1°特征峰和Ⅱ晶型在12.9°特征峰来确定标准曲线(如图4,图中relative intensity为Ⅱ晶型特征峰强度与两 种晶型峰强度之和的比值)。
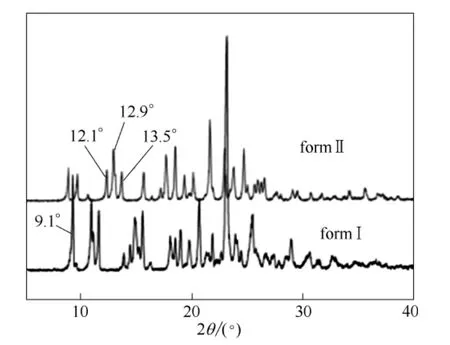
图3 硫酸氢氯吡格雷不同晶型的特征峰 Fig.3 Characteristic peaks selected for different forms of CHS
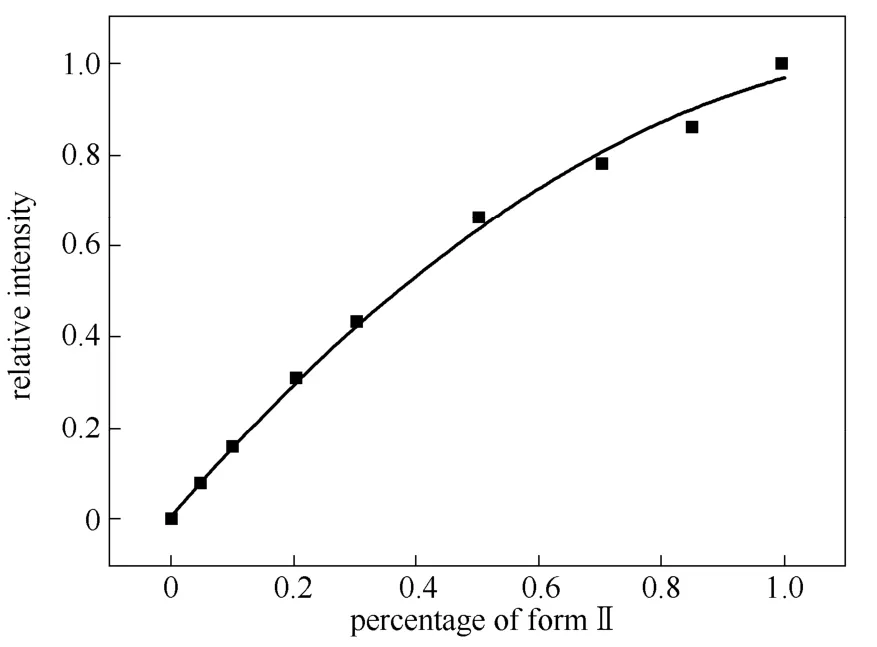
图4 Ⅱ晶型特征峰相对强度与实际含量的关系 Fig.4 Relative intensity of characteristic peaks vsCHS form Ⅱ percentage
根据上面所得的标准曲线,针对水和二氯甲烷对晶型的纯度影响进行了测量,得到的结果分别如图5和图6所示(图中percent指根据上述标准曲线所得的Ⅱ晶型在产品中的比例)。
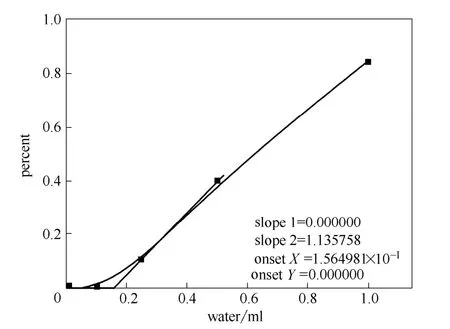
图5 水含量对晶型的影响 Fig.5 Effect of water on polymorph of CHS
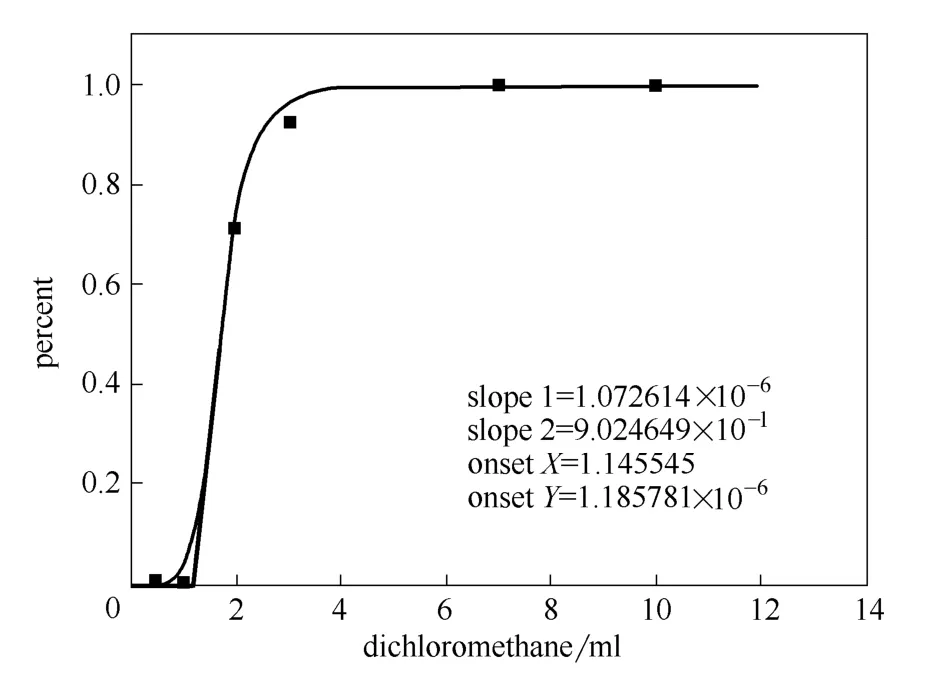
图6 二氯甲烷含量对晶型的影响 Fig.6 Effect of dichloromethane on polymorph of CHS
实验结果表明,当产品中出现Ⅱ晶型时,水分的含量为0.156 ml,而二氯甲烷的含量则为1.14 ml,并且当杂质溶剂的含量增加时,Ⅱ晶型的含量迅速增加,直至全部变为Ⅱ晶型。因此残留溶剂的存在会加速Ⅱ晶型的形成,残留水分对晶型的影响更大。
2.3 球形晶体的表征
2.3.1 扫描电镜照片对比 对实验所得的球形产品与一般的非球形产品做对比,其中扫描电镜(SEM)的照片对比如图7所示。不难看出,球形产品粒度明显高于非球形产品,且产品粒度分布均匀。
2.3.2 粒度分布对比 用Mastersizer3000测定球形与非球形晶体的粒度分布(图8),在测定过程中,非球形产品聚结严重,不能分散在溶剂中,因此, 在测定粒度分布之前,首先用超声使非球形产品分散开。

图7 二氯甲烷含量对晶型的影响 Fig.7 SEM pictures for spherical and non-spherical products of CHS (form Ⅰ)
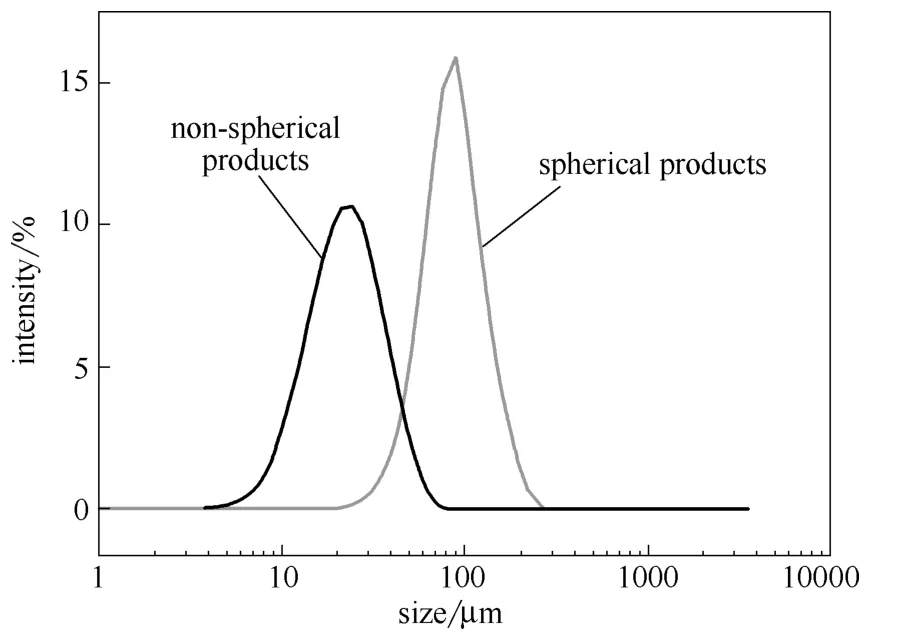
图8 硫酸氢氯吡格雷Ⅰ晶型球形晶体与非球形晶体粒度 分布对比 Fig.8 Particle size distribution for spherical and non-spherical products of CHS (form Ⅰ)
从图中可以看出,球形产品的粒度明显要大于非球形产品,而其粒度分布范围也较非球形产品要稍窄,粒度分布更为均一。而通过两种产品Span值的对比则能更加直观地看出其粒度分布的区别。Span值由下式得到
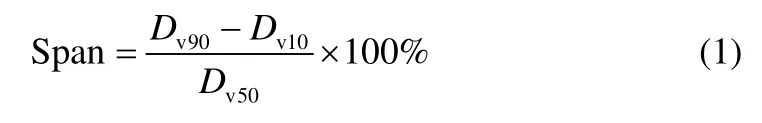
式中,Dv90、Dv50、Dv10分别代表粒子所占体积分数小于90%,50%,10%的粒子粒径。
Span值越小,表明粒度分布越窄。由式(1)计算得,非球形产品的Span值为4.81,而球形晶体的Span值仅为1.79,同样说明了球形晶体粒度分布更加均一。
2.3.3 堆密度对比 堆密度与振实密度是评价晶体的重要指标。只有振实密度达到一定水平后,才能直接压片,另外,堆密度的提高还可有利于产品的储存与运输情况。
球形产品与非球形产品密度比较见表1。
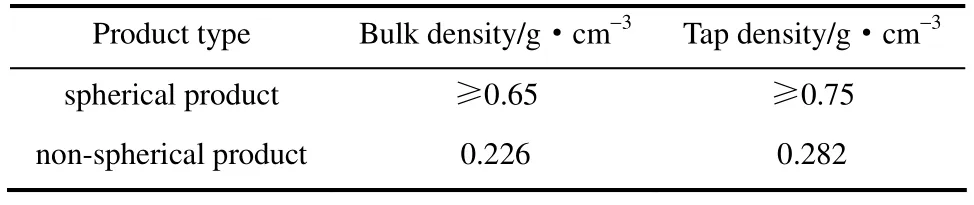
表1 球形与非球形产品堆密度对比 Table 1 Density comparison for spherical and non-spherical products
2.3.4 晶体稳定性对比 已有文献报道球形的晶体可抑制晶体的转变,而对于硫酸氢氯吡格雷晶体,由于目标产品为介稳晶型,因此,球形晶体有利于产品的稳定。
利用红外监控转晶过程中不同晶型的浓度,通过浓度变化表征晶型的转化过程,发现如图9显示的现象:非球形产品在实验条件下大约经过2 h,在浓度上会存在一个大的阶梯状下降,而球形的产品浓度变化则延长到5 h。由此可见,球形产品对于晶体的转晶也存在抑制作用。
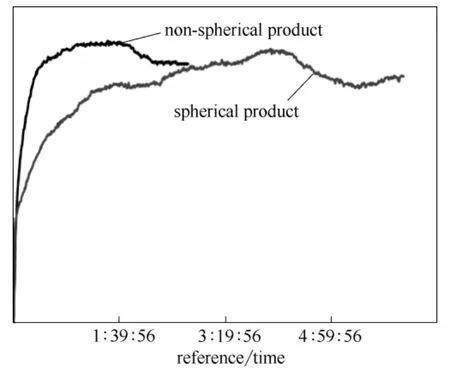
图9 球形与非球形产品转晶速率的比较 Fig.9 Transformation rate of spherical and non-spherical products of CHS (form Ⅰ)
2.4 球形晶体生长机理
实验过程中,利用显微镜和扫描电镜观察过程中晶习,实验结果如图10所示。
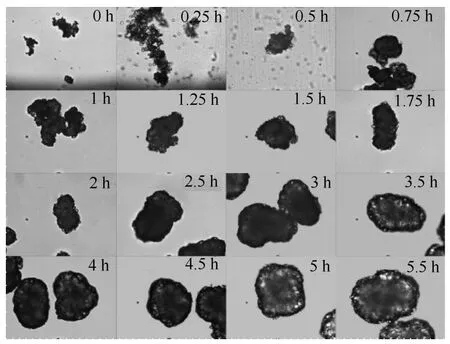
图10 硫酸氢氯吡格雷球晶生长过程 Fig.10 Growth process of CHS spherical particles
实验过程中,显微镜观察发现,硫酸氢氯吡格雷Ⅰ晶型成球的机理与一般的球形结晶不同。一般 情况下,产品成球是先生成单晶,然后在某种因素的影响下,单晶间发生聚结,最终聚结成球形颗粒。但硫酸氢氯吡格雷Ⅰ晶型的球形颗粒并不是由生成的单晶聚结而成,而是直接生长形成的多晶。成球的机理应为球形生长机理。实验中,通过不同时间点处取样观察了晶体生长的过程(图10)。
从实验过程开始至实验结束,都没有观察到单晶的出现。在加入硫酸后,相当长的时间内溶液中都无法自发成核,只有加入晶种诱导产品析出。由于溶液中存在很高的过饱和度,在晶种的诱导下,马上成核,原本分散、单一的晶体晶种周围产生很多晶体,形成粒状。但此时的晶体是松散且连在一起的。这与硫酸氢氯吡格雷容易聚结的性质有关。随着晶体的生长,晶种逐渐分散开,作为独立的核。当实验进行到0.75 h时,产品已变得比较紧实。不过此时的颗粒还是堆积在一起,到1.25 h时,颗粒分散开,独立生长直至最终生长成为分散的大颗粒球形晶体。实验起初,颗粒的形状还不是球形,颗粒的轮廓处还不是很光滑,存在很多的夹角,但随着时间增长,结晶过程的进行,颗粒逐渐长大并且球形度逐渐增高,最终得到目标产品。当实验进行到1.75 h时,颗粒的表面凹陷或突出的部分已经基本消失了。在晶体生长过程中,在颗粒上有凹陷的地方晶体的生长速率更快,最终产品变成球状。通过图10中的照片可以推测,球形颗粒的形成是一个生长的过程,而不是一个聚结的过程。抽取实验过程中的样品,可以对上述推论进行验证。中间取样2 h产品,利用电子显微镜观察球形颗粒表面。其结果如图11所示。
从图中看出,颗粒的表面并不是光滑的,有很大的缝隙,且表面上的晶体呈向外辐射状,与最终产品表面光滑且缝隙小的特点不一致,并且可清楚 地看到:晶体簇是在内层表面上生长的。并且经粒度分析可知,2 h的产品粒径要小于最终的产品。

图11 过程取样(2 h)SEM照片 Fig.11 SEM pictures of sampling at 2 h during CHS spherical crystallization
硫酸氢氯吡格雷Ⅰ晶型球形晶体的形成机理:首先在晶种的表面发生2D成核,然后,溶质分子在晶体的表面,顺着晶核,沿着外层表面开始生长,逐渐将颗粒的表面填满,然后,又会在已填满的一层上晶体又成核,继续生长,将晶核间的空隙填满,直至最终生成球形晶体。而最终球形晶体的大小可能与传质、流场等因素有关。球晶生成机理如图12所示。
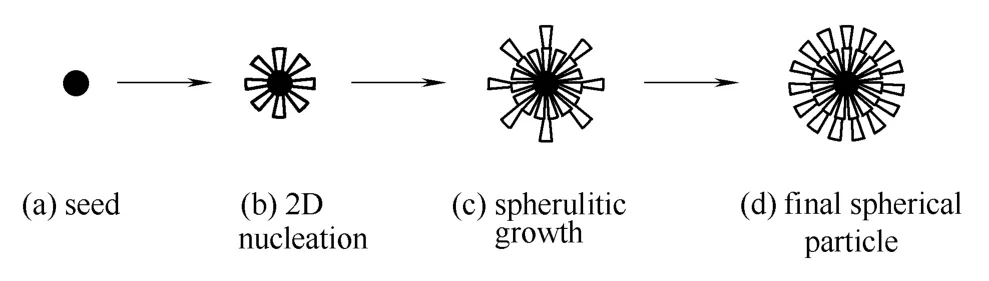
图12 球形晶体形成示意图 Fig.12 Sketch for CHS spherulitic growth process
3 结 论
(1)硫酸氢氯吡格雷Ⅰ、Ⅱ晶型在仲丁醇中的溶解度随温度升高而升高,但两种晶型溶解度差随温度变化不大,高温更有利于介稳晶型向稳定晶型的转化。
(2)对比球形产品与非球形产品在堆密度、粒度分布和晶体稳定性方面的差异,发现球形晶体不仅堆密度远高于非球形晶体,利于后续压片操作,且晶体的稳定性也得到了提高,更利于产品后期的储存等操作。
(3)通过过程取样分析和研究,发现硫酸氢氯吡格雷Ⅰ晶型球形产品并非由细晶聚结而成,而是由晶种表面的2D成核以及之后的球形生长得到,探明了硫酸氢氯吡格雷Ⅰ晶型球形晶体的生长机理。
符 号 说 明
Dv90——粒子所占体积分数小于90%的粒子粒径
Dv50——粒子所占体积分数小于50%的粒子粒径
Dv10——粒子所占体积分数小于10%的粒子粒径
[1] Mangin D, Puel F, Veesler S.Polymorphism in processes of crystallization in solution: a practical review [J].Organic Process Research & Development, 2009, 13 (6): 1241-1253.
[2] Sharma P, Zujovic Z D, Bowmaker G A, Denny W A, Garg S.Evaluation of a crystalline nanosuspension: polymorphism, process induced transformation and in vivostudies [J].International Journal of Pharmaceutics, 2011, 408 (1): 138-151.
[3] Jiang Shanfeng, Jansens P J, ter Horst J H.Control over polymorph formation of o-aminobenzoic acid [J].Crystal Growth & Design, 2010, 10 (6): 2541-2547.
[4] Martínez-Ohárriz M C, Martin C, Goni M M, Rodríguez- Espinosa C, Ilarduya-Apaolaza D, Tros M C, Sánchez M.Polymorphism of diflunisal: isolation and solid-state characteristics of a new crystal form [J].Journal of Pharmaceutical Sciences, 1994, 83 (2): 174-177.
[5] Kralj D, Brečević L, Nielsen A E.Vaterite growth and dissolution in aqueous solution (Ⅰ): Kinetics of crystal growth [J].Journal of Crystal Growth, 1990, 104 (4): 793- 800.
[6] Jiang Shanfeng, Jansens P J, ter Horst J H.Mechanism and kinetics of the polymorphic transformation of o- aminobenzoic acid [J].Crystal Growth & Design, 2010, 10 (5): 2123-2128.
[7] Nath N K, Aggarwal H, Nangia A.Crystal structure of methyl paraben polymorph Ⅱ [J].Crystal Growth & Design, 2011, 11 (4): 967-971.
[8] Roy S, Bhatt P M, Nangia A, Kruger G J.Stable polymorph of venlafaxine hydrochloride by solid-to-solid phase transition at high temperature [J].Crystal Growth & Design, 2007, 7 (3): 476-480.
[9] Mo Yuxin, Dang Leping, Wei Hongyuan.L-Glutamic acid polymorph control using amino acid additives [J].Industrial & Engineering Chemistry Research, 2011, 50 (18): 10385-10392.
[10] Price C P, Grzesiak A L, Matzger A J.Crystalline polymorph selection and discovery with polymer heteronuclei [J].Journal of the American Chemical Society, 2005, 127 (15): 5512-5517.
[11] McKellar S C, Urquhart A J, Lamprou D A, Florence A J.Polymer templating of supercooled indomethacin for polymorph selection [J].ACS Combinatorial Science, 2012, 14 (3): 155-159.
[12] Croker D, Hodnett B K.Mechanistic features of polymorphic transformations: the role of surfaces [J].Crystal Growth & Design, 2010, 10 (6): 2806-2816.
[13] Doki N, Seki H, Takano K, Asatani H, Yokota M, Kubota N.Process control of seeded batch cooling crystallization of the metastable α-form glycine using an in-situATR- FTIR spectrometer and an in-situFBRM particle counter [J].Crystal Growth & Design, 2004, 4 (5): 949-953.
[14] Hausman D S, Cambron R T, Sakr A.Application of on-line Raman spectroscopy for characterizing relationships between drug hydration state and tablet physical stability [J].International Journal of Pharmaceutics, 2005, 299 (1): 19-33.
[15] Braun D E, Karamertzanis P G, Arlin J B, Florence A J, Kahlenberg V, Tocher D A, Price S L.Solid-state forms of β-resorcylic acid: how exhaustive should a polymorph screen be? [J].Crystal Growth & Design, 2010, 11 (1): 210-220.
[16] Kawashima Y, Okumura M, Takenaka H.Spherical crystallization: direct spherical agglomeration of salicylic acid crystals during crystallization [J].Science, 1982, 216 (4550): 1127-1128.
[17] Zhang Haitao, Chen Ying, Wang Jingkang, Gong Junbo.Investigation on the spherical crystallization process of cefotaxime sodium [J].Industrial & Engineering Chemistry Research, 2009, 49 (3): 1402-1411.
[18] Kawashima Y, Aoki S, Takenaka H, Miyake Y.Preparation of spherically agglomerated crystals of aminophylline [J].Journal of Pharmaceutical Sciences, 1984, 73 (10): 1407-1410.
[19] Kawashima Y, Niwa T, Handa T, Takeuchi H, Iwamoto T, Itoh K.Preparation of controlled-release microspheres of ibuprofen with acrylic polymers by a novel quasi-emulsion solvent diffusion method [J].Journal of Pharmaceutical Sciences, 1989, 78 (1): 68-72.
[20] Nocent M, Bertocchi L, Espitalier F, Baron M, Couarraze G.Definition of a solvent system for spherical crystallization of salbutamol sulfate by quasi-emulsion solvent diffusion (QESD) method [J].Journal of Pharmaceutical Sciences, 2001, 90 (10): 1620-1627.
[21] Ueda M, Nakamura M Y, Makita H, Imasato Y, Kawashima Y.Particle design of enoxacin by spherical crystallization technique [J].Chem.Pharm.Bull., 1990, 38 (9): 2537-2541.
[22] Puechagut H G, Bianchotti J, Chiale C A.Preparation of norfloxacin spherical agglomerates using the ammonia diffusion system [J].Journal of Pharmaceutical Sciences, 1998, 87 (4): 519-523.
[23] Pawar A P, Paradkar A R, Kadam S S, Mahadik K R.Crystallo-co-agglomeration: a novel technique to obtain ibuprofen- paracetamol agglomerates [J].AAPS Pharm.Sci.Tech., 2004, 5 (3): 57-64.
[24] Kawashima Y, Handa T, Takeuchi H.Crystal modification of phenytoin with polyethylene glycol for improving mechanical strength, dissolution rate and bioavailability by a spherical crystallization technique [J].Chemical & Pharmaceutical Bulletin, 1986, 34 (8): 3376-3383.
