气凝胶骨架镶嵌的CeO2纳米团簇催化氧化HCl制Cl2
2015-08-20徐希化费兆阳陈献汤吉海崔咪芬乔旭
徐希化,费兆阳,陈献,汤吉海,崔咪芬,乔旭,
(1 南京工业大学材料化学工程国家重点实验室,江苏 南京 210009;2 南京工业大学化工学院,江苏 南京 210009)
引 言
Cl2作为一种重要的基础原料广泛应用于许多化学工业过程中,尤其是快速增长的聚氨酯、聚碳酸酯行业,然而这些反应过程副产大量的HCl,会导致氯资源的浪费和严重的环境污染[1]。以聚氨酯中间体甲苯二异氰酸酯的生产为例[2-3],所有的氯原子均未出现在最终的产品中,全部以HCl 的形式排出。催化氧化法将HCl 制成Cl2以实现氯资源的循环利用,是实现涉氯化学工业绿色生产和节能减排的重要途径[4-6]。
CeO2可以快速地进行氧化还原循环,具有很好的储释氧性能,被广泛用作催化剂载体、活性组分或者助剂[7-9]。CeO2具有良好的HCl 催化氧化活性和稳定性,被认为是有希望的新型HCl 氧化催化剂[10-11]。研究认为CeO2的HCl 氧化催化性能与其表面的氧空位浓度密切相关[12]。通过对CeO2进行过渡金属掺杂,可以提升CeO2的氧空位浓度、氧化还原能力和热稳定性[13-15],但在HCl 氧化过程中金属掺杂的CeO2容易发生表面重构,掺杂元素从固溶体中析出而使催化剂稳定性较差。通过调节载体性质和制备方法,获得CeO2高度分散负载型催化材料,也是提高其催化性能的有效手段[16]。但是常规技术制备的负载型催化剂, CeO2分散容量较低,导致催化效率提升仍然有限。
本文通过一步溶胶-凝胶法合成了一系列镶嵌于气凝胶骨架内的 CeO2纳米团簇催化材料(CeO2@MxOy,MxOy=SiO2、Al2O3、ZrO2)。利用ICP、XRD、SEM、N2吸附-脱附、H2-TPR 和OSC(储氧量)等表征考察了催化剂的结构,并系统考察其催化HCl 氧化制Cl2的性能。在此基础上,通过动力学对表面反应过程进行初步探究。
1 实验部分
1.1 实验材料
六水合硝酸铈(Ce(NO3)3·6H2O,AR)、九水合硝酸铝(Al(NO3)3·9H2O,AR)、环氧丙烷(C3H6O,AR)、甲酰胺(HCONH2,AR)、冰乙酸(CH3COOH,AR)和正硅酸四乙酯(TEOS,AR)购自国药集团化学试剂有限公司;硝酸氧锆(ZrO(NO3)2,AR)购自阿拉丁试剂;无水乙醇(CH3CH2OH,AR)购自无锡市亚盛化工有限公司;氧气(O2,纯度为99.9%)购自南京天泽气体有限公司;氮气(N2,纯度为99.9%)、HCl(纯度为99.8%)购自南京三乐集团有限公司。
1.2 催化剂制备
催化剂采用一步溶胶-凝胶法制备。以CeO2@SiO2的制备为例, 称取一定量的Ce(NO3)3·6H2O 和TEOS 于无水乙醇和蒸馏水的混合溶剂中(前驱体物质的量总和为0.1 mol),搅拌至完全溶解,在继续搅拌的同时,依次加入3.1 g乙酸、4.5 g 甲酰胺、70.0 g 环氧丙烷。在70℃水浴中进行凝胶和老化 6 h,再在室温下进一步老化12 h。用无水乙醇洗涤后,再用15%体积含量的TEOS/乙醇溶液浸泡凝胶48 h 改性处理,用无水乙醇浸泡48 h 得到湿凝胶,再将得到的湿凝胶在60℃真空干燥50 h,最后在500℃空气氛围中焙烧3 h,即制得所需样品。xCeO2@SiO2,其中 x 代表投料中100×nCe/(nCe+nSi)。相同的制备方法得到CeO2@ZrO2和CeO2@Al2O3。
1.3 催化剂表征
ICP 通过Optima2000DV 检测催化剂中Ce 元素量,并计算出CeO2质量含量;XRD 采用Rigaku 公司(日本)的SmartLab 衍射仪进行检测,利用CuKα为射线源(λ = 0.154 nm),管电流为100 mA,电压为40 kV,扫描范围2θ = 10°~80°。催化剂的晶粒尺寸和晶胞参数由Scherrer 和Bragg 公式计算得到;N2吸附-脱附测试使用 BEL 公司(日本)的BELSORP-Ⅱ型吸附仪对制备的催化剂进行测试,用BET 方程计算样品的比表面积,并使用BJH 模型计算样品的孔容和孔径分布;SEM 是在FEI 公司(荷兰)的QUANTA-200 型环境扫描电镜上进行的;H2-TPR 和OSC(储氧量)均在Micromeritics公司(美国)的Auto Chem 2920 仪器上对样品进行测试。
1.4 催化性能评价
采用内径为24 mm 的石英管作为反应器,将催化剂与石英砂以1:10 的比例混合后装填在反应管的中段。通过固定床的三段加热炉来控制反应段催化剂的床层温度,通过质量流量计来控制反应气体HCl 和氧气的流量。混合气体由反应器的顶部进入,从反应器的底部排出,后通过缓冲瓶,再通过2 个碱液吸收瓶中和处理后排空。反应系统底部设三通阀,用于取样分析。
催化剂动力学测试过程中限制HCl 转化率在5%~20%,催化剂用量为1 g,以N2作为平衡气,保证总气体进料流速为300 ml·min-1。
反应产物用KI 溶液进行吸收后,用碘量法及酸碱滴定法测定生成的Cl2及未反应的HCl,计算出HCl 的转化率(XHCl)
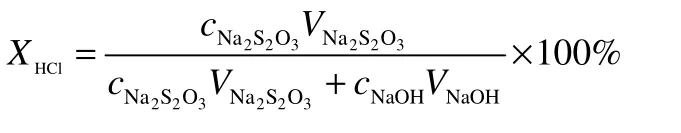
2 实验结果与讨论
2.1 XRD 分析
图1 是CeO2@MxOy催化剂的XRD 谱图。从图1 可以看到,所有样品均未能明显观察到对应于基底氧化物的特征峰,这说明基底氧化物以非晶的形式存在。除20CeO2@ZrO2外,其他样品均能观察到对应于立方萤石结构的CeO2特征峰[17],但其峰强度均较弱且高度弥散,这说明CeO2以较小的晶粒尺寸存在。在CeO2@ZrO2和CeO2@Al2O3催化剂中,CeO2的特征峰峰强度随铈含量的增加而略有提升。CeO2@SiO2中CeO2的特征峰峰强度较小且随铈含量的增加没有明显的变化,这表明CeO2始终能以很小的晶粒尺寸存在于基底SiO2骨架结构中。
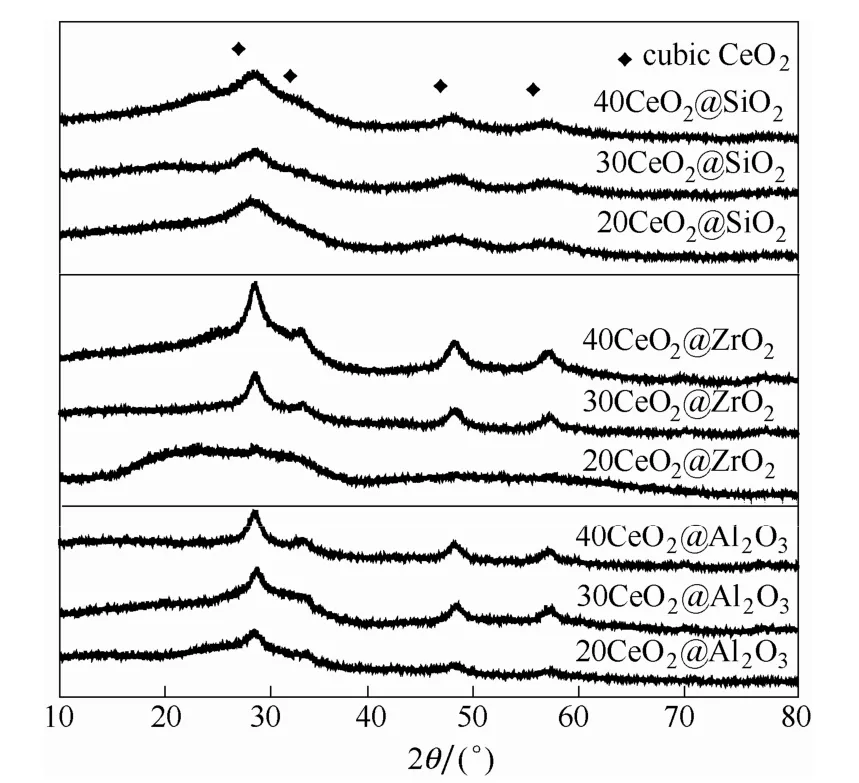
图1 CeO2@MxOy 催化剂的XRD 谱图Fig.1 XRD patterns of CeO2@MxOy catalysts
2.2 N2 吸附-脱附分析
图2 是CeO2@MxOy催化剂的N2吸附-脱附等温线。从图2 可以看到,各催化剂的等温线虽有所差别,但均为Ⅳ型等温线,表明所有的样品均具有介孔结构。除40CeO2@Al2O3外,所有样品均具备气凝胶典型的H2 型回滞环,这是一种典型的“墨水瓶”孔结构[18],这表明催化剂孔道主要由纳米级别的三维网络骨架构成。40CeO2@Al2O3则呈现出H3 型回滞环,这表明其催化剂孔道不规则,这可能是气凝胶骨架发生破坏从而形成了较多的堆积 孔结构。
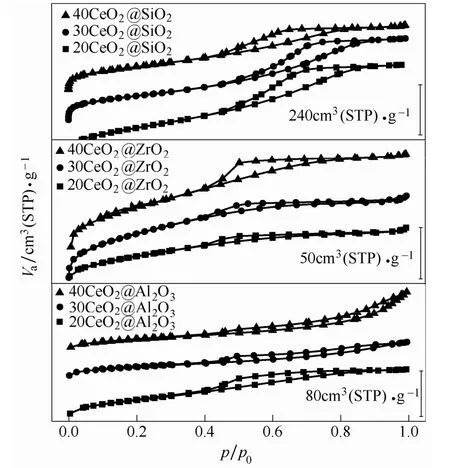
图2 CeO2@MxOy 催化剂的N2 吸脱附等温线Fig.2 N2 Adsorption-desorption isotherms of CeO2@MxOy catalysts
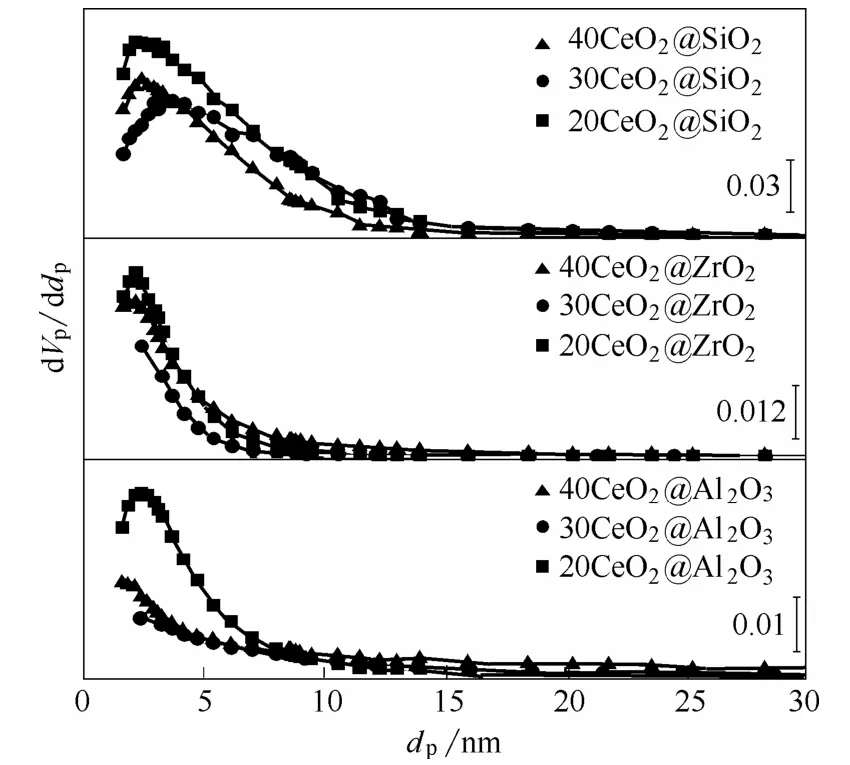
图3 CeO2@MxOy 催化剂的孔径分布图Fig.3 Pore size distributions of CeO2@MxOy catalysts
进一步利用BJH 方法计算得到的CeO2@MxOy催化剂的孔径分布(图3)。从图3 可以看到,CeO2@ZrO2和CeO2@Al2O3的孔径主要集中在2~10 nm 范围内,而CeO2@SiO2孔径在2~15 nm 的较宽范围内,这说明CeO2@SiO2更容易形成较大的孔结构。催化剂的比表面积和孔体积数据列于表1。从表1 可以看出,CeO2@SiO2具有较高的比表面积(473.5 ~687.2 m2·g-1)和 孔 容(0.5055 ~0.7573 cm3·g-1)。而CeO2@ZrO2和CeO2@Al2O3的比表面积和孔容较小,这可能是由于气凝胶骨架发生严重破坏从而形成较多的堆积孔结构,从而导致更小的比表面积和孔容[19]。
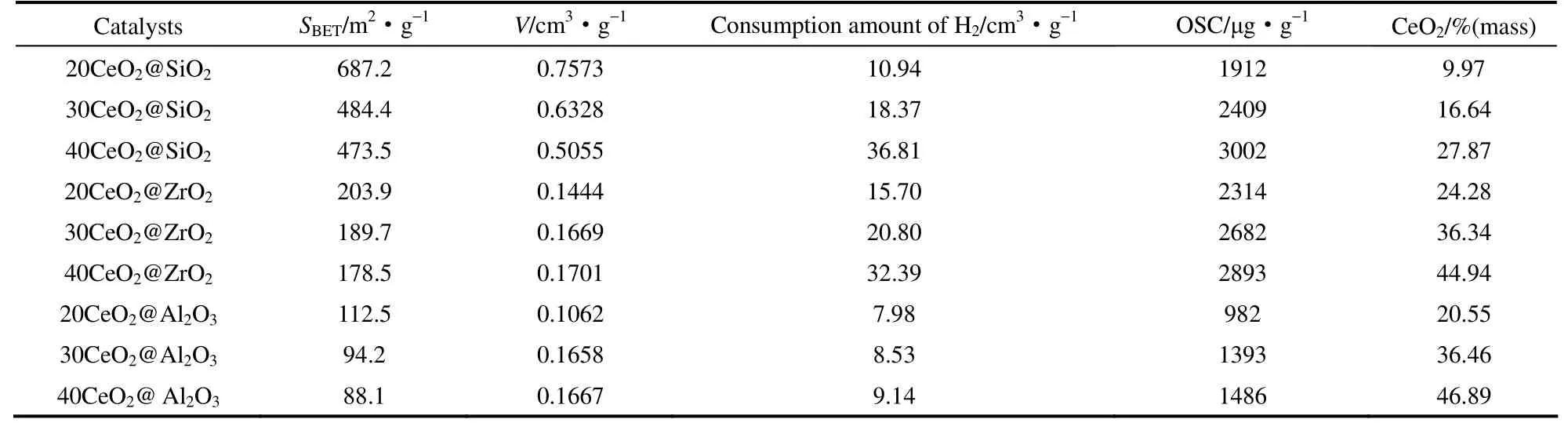
表1 CeO2@MxOy 催化剂的物理化学性质Table1 Physical chemical properties of CeO2@MxOy catalysts
2.3 SEM 分析
图4 是CeO2@MxOy催化剂的SEM 和EDX 图。所有催化剂的整体形貌均呈现出块状结构[图4(a)、(e)和(i)]。从图4(b)、(f)和(j)可以看出,3 种催化剂均形成均匀的颗粒。其中,40CeO2@SiO2[图(b)]的颗粒尺寸最小且排列紧密,而40CeO2@ZrO2[图(f)]和 40CeO2@Al2O3[ 图(j)]颗 粒 尺 寸 接 近, 但40CeO2@Al2O3的颗粒排列较为松散,这有可能导致其气凝胶骨架发生破坏,从而降低了其比表面积和孔容。另外,各催化剂的元素分析图显示Si、Zr、Al 及Ce 元素分散均匀,这表明CeO2粒子可以相对均匀地分散在不同的气凝胶骨架内。

图4 (a)~(d) 40CeO2@SiO2, (e)~(h) 40CeO2@ZrO2 和(i)~(l) 40CeO2@Al2O3 催化剂的SEM 和EDX 图Fig.4 SEM and EDX images of 40CeO2@SiO2[(a)—(d)], 40CeO2@ZrO2 [(e)—(h)]and 40CeO2@Al2O3 [(i)—(l)]catalysts
2.4 H2-TPR 和OSC 分析
图5 是CeO2@MxOy催化剂的H2-TPR 结果。因为SiO2、Al2O3和ZrO2在测试温区内很难被还原,所以还原峰主要为CeO2的还原。通常CeO2在低于600℃的还原峰为其表面氧物种的还原,而在高于600℃的还原峰为其体相氧的还原[20]。在本文所有CeO2@MxOy的还原曲线中只在300~600℃之间出现较宽的低温还原峰,未出现体相CeO2的高温还原峰,这与晶相CeO2的还原性质有很大不同,这可能是由于一步溶胶-凝胶法所制备的样品中CeO2均以很小尺寸的纳米团簇分散于气凝胶骨架中造成的。各催化剂的还原耗氢量结果列于表1。从表1可以看出,CeO2@SiO2和CeO2@ZrO2均显示出较高的还原耗氢量,其中40CeO2@SiO2最高可达36.81 cm3·g-1。
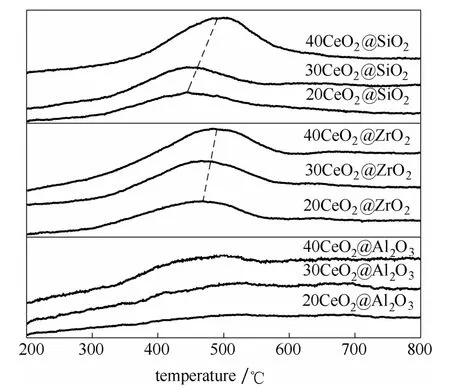
图5 CeO2@MxOy 催化剂的H2-TPR 图Fig.5 H2-TPR profile of CeO2@MxOy catalysts
CeO2@MxOy催化剂的OSC 结果列于表1。从表1 可以看到,CeO2@MxOy中不同基底对CeO2有显著不同的影响。CeO2@SiO2和CeO2@ZrO2显示出更高的储释氧能力,且储氧量随着CeO2含量的增加而显著提高,40CeO2@SiO2和40CeO2@ZrO2储氧量分别高达3002 和2893 μg·g-1。CeO2@Al2O3储氧量均较低,最高的40CeO2@Al2O3为1486 μg·g-1,约为40CeO2@ZrO2和40CeO2@SiO2储氧量的一半。CeO2纳米团簇的量子尺寸效应导致各催化剂的储氧量均远高于文献报道的纯CeO2的储氧量[10,21]。
2.5 催化剂的反应性能测试
2.5.1 催化剂活性测试 图6 是不同CeO2质量含量的CeO2@MxOy催化剂在430℃下的活性测试,CeO2质量含量由ICP 测得,结果列于表1。从图6可以看到,CeO2@SiO2、CeO2@ZrO2和CeO2@Al2O3均显示出较高的HCl 氧化催化活性,并随着CeO2含量的增加而有所提升。在氧氯比为2、HCl 与催化剂接触时间(t)为0.3196 h、反应温度为430℃时,40CeO2@SiO2和40CeO2@ZrO2催化剂上Cl2的空时产率分别达到1.60 和1.48 g·g-1·h-1。
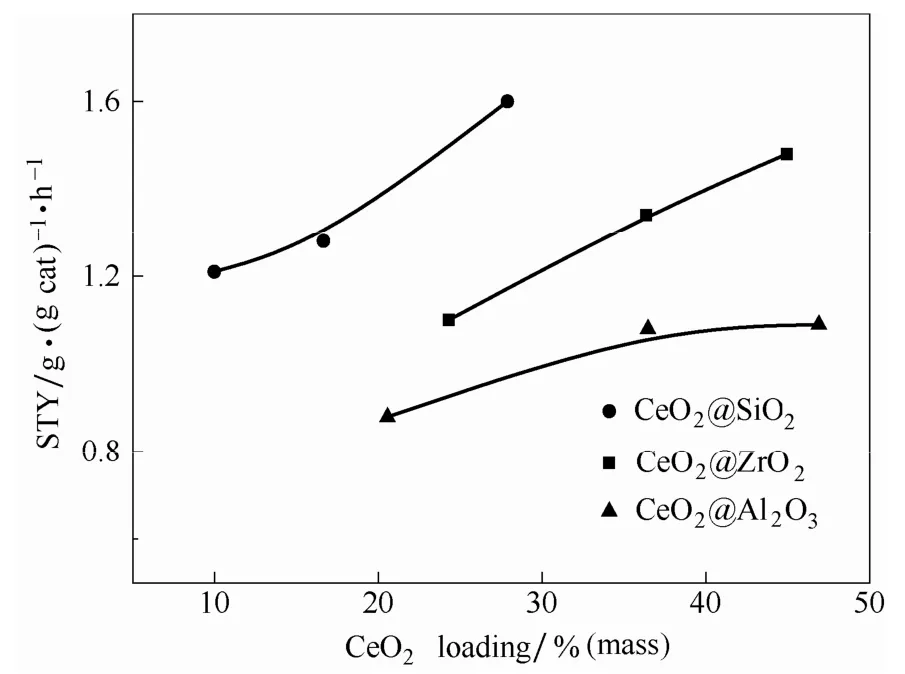
图6 CeO2@MxOy 催化剂CeO2 含量与催化活性关系图Fig.6 Catalytic activity with different CeO2 loading of CeO2@MxOy catalysts
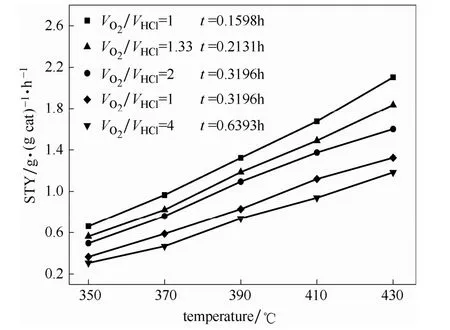
图7 40CeO2@SiO2 催化剂在不同条件下的活性测试Fig.7 Activity tests of 40CeO2@SiO2 catalysts at various conditions
图7是催化活性最高的40CeO2@SiO2催化剂在 不同条件下的工艺考察结果。从图7 可以看出,40CeO2@SiO2催化剂在较低温度下的活性差别不大,当反应温度升高时,催化剂在不同测试条件下表现出的活性差异随之变大。当VO2/VHCl增加,或者HCl 与催化剂接触时间减少,40CeO2@SiO2的Cl2空时产率随之增加。在VO2/VHCl为1、HCl 与催化剂接触时间为 0.1598 h、430 ℃条件下,40CeO2@SiO2的 Cl2空时产率可以达到 2.10 g·g-1·h-1。将40CeO2@SiO2催化剂的HCl 氧化催化性能与文献所报道的结果进行了对比,控制本文的实验条件与文献中的条件类似。由表2 中的数据可以看到,40CeO2@SiO2催化剂较低的氧氯比下,表现出了较其他催化剂明显更好的HCl 氧化催化活性。
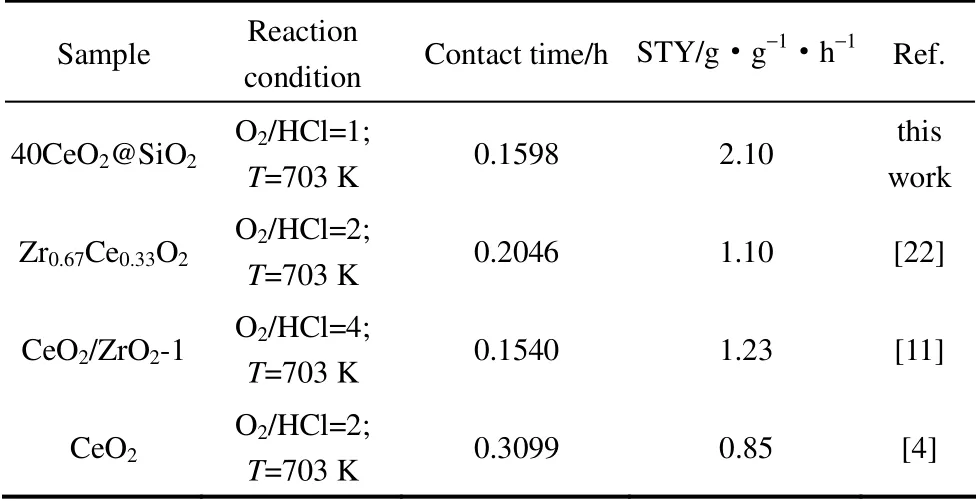
表2 CeO2@SiO2HCl 氧化催化活性与文献报道的比较Table 2 Comparison of HCl oxidation over CeO2@SiO2 with catalysts reported in literatures
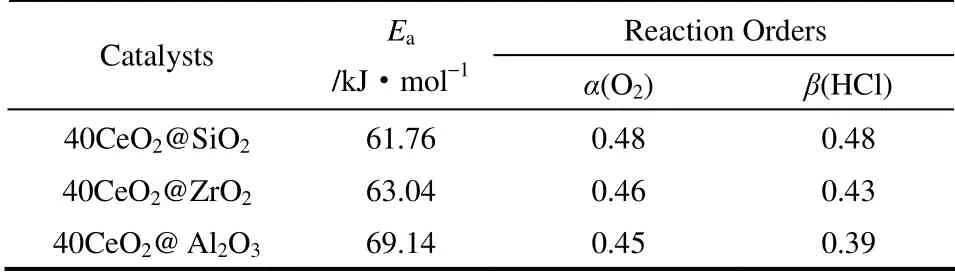
表3 CeO2@MxOy 催化剂活化能和反应级数测定Table 3 Reaction orders and activation energies (Ea) for CeO2@SiO2, CeO2@Al2O3 and CeO2@ZrO2 catalysts
2.5.2 催化剂动力学测试与机理分析 40CeO2@MxOy催化剂的表观活化能通过Arrhenius方程和速率方程计算得到,列于表3。从表3 可以看到,40CeO2@MxOy催化剂的表观活化能均在60~70 kJ·mol-1之间,且比较接近,说明这3 种催化剂的反应机理相同。最高活性的40CeO2@SiO2的表观活化能较低,仅为61.76 kJ·mol-1。这可能由于40CeO2@SiO2催化剂中CeO2较小尺寸的纳米团簇使反应物HCl 和氧气更易于在催化剂表面的吸附和活化,从而促进了HCl 氧化反应的快速进行。各催化剂对HCl 和氧气的反应级数采用微分法测得[23],结果列于表3。分别增加O2和HCl 分压,Cl2的产率都是增加的,这说明O2或者HCl 的吸附以及表面反应不可能是反应的决速步骤。同时反应过程中所生成的Cl2首先会吸附在催化剂的表面,O2和HCl 作为反应物与Cl2在催化剂表面会产生竞争吸附[12],从而可推断Cl2在催化剂上的脱附是该反应的决速步骤。
3 结 论
(1)采用一步溶胶-凝胶法制备的一系列CeO2@MxOy(MxOy=SiO2、ZrO2、Al2O3)催化剂。表征结果显示,CeO2以纳米团簇镶嵌于气凝胶骨架内,量子尺寸效应导致其在晶相CeO2表面氧物种还原温区内即可被充分还原,表现出完全不同于晶相CeO2的氧化还原性能。其中,40CeO2@SiO2具有最大的比表面积,最强的氧化还原能力和储释氧能力。
(2)CeO2@MxOy在HCl 催化氧化过程具有较高的活性,其中,40CeO2@SiO2的活性最好,在HCl 接触时间为0.1598 h 、氧氯比为1、反应温度430℃时,催化剂的Cl2空时产率可以达到2.10 g·g-1·h-1。动力学测试结果显示,催化剂表面的HCl 氧化反应同时受O2分压和HCl 分压的影响,这表明Cl2从催化剂表面的脱附是该反应的决速步骤。
[1]Mortensen M K, Minet R G, Tsotsis T T, Benson S.A two-stage process for catalytic oxidation of hydrogen chloride to chlorine [J].Chemical Engineering Science, 1996, 51(10):2031-2039.
[2]Wu Liding(吴礼定).Production presents status of TDI and its research development [J].China Chlor-Alkali(中国氯碱), 2011, (10): 19-21.
[3]Zhang Linhao(张林浩), Liang Rizhong(梁日忠).Eco-industry mode study on the process of polyurethane intermediate production with chlorine element recycle [J].Chemical Industry and Engineering Progress(化工进展), 2012, 30(12): 2799-2803.
[4]Amrute A P, Mondelli C, Hevia M A G, Pérez-Ramírez J.Mechanism-performance relationships of metal oxides in catalyzed HCl oxidation [J].ACS Catalysis, 2011, 1(6): 583-590.
[5]Tang J H, Chen X, Fei Z Y, Zhao J H, Cui M F, Qiao X.HCl oxidation to recycle Cl2over a Cu/Ce composite oxide catalyst(Ⅰ): Interinsic kinetic study [J].Industrial & Engineering Chemistry Research, 2013, 52(34): 11897-11903.
[6]Pérez-Ramírez J, Mondelli C, Schmidt T, Schlüter O F K, Wolf A, Mleczko L, Dreier T.Sustainable chlorine recycling via catalysed HCl oxidation: from fundamentals to implementation [J].Energy & Environmental Science, 2011, 4(12): 4786.
[7]Li H L, Wu C Y, Li L Q, Li Y, Zhao Y C, Zhang J Y.Kinetic modeling of mercury oxidation by chlorine over CeO2-TiO2catalysts [J].Fuel, 2013, 113:726-732.
[8]Yin Fengxiang(银凤翔), Ji Shengfu(季生福), Chen Nengzhan(陈能展), Zhao Liping(赵丽萍), Wang Wei(王伟), Li Chengyue(李成岳), Liu Hui(刘辉).Catalytic combustion of methane over Ce1-xCuxO2-x/ Al2O3solid solution catalyst [J].Journal of Chemical Industry and Engineering (China) (化工学报), 2006, 57(4): 744-750.
[9]Meng Yingqin(孟英芹), Zhao Xiaobing(赵晓兵), Li Xiazhang(李霞章).Catalytic properties of CeO2-TiO2/attapulgite nanocomposites prepared by sol-gel method [J].CIESC Journal(化工学报), 2013, 64(7): 2679-2686.
[10]Amrute A P, Mondelli C, Moser M, Novell-Leruth G, Lopez N, Rosenthal D, Farr R, Schuster M E, Teschner D, Shmidt T, Pérez-Ramírez J.Performance, structure, and mechanism of CeO2in HCl oxidation to Cl2[J].Journal of Catalysis, 2012, 286: 287-297.
[11]Moser M, Mondelli C, Schmidt T, Girgsdies F, Schuster M E, Farra R, Szentmiklosi L, Tesschner D, Pérez-Ramírez J.Supported CeO2catalysts in technical form for sustainable chlorine production [J].Applied Catalysis B, 2013, 132/133: 123-131.
[12]Fei Z Y, Xie X X, Dai Y, Liu H Y, Chen X, Tang J H, Cui M F, Qiao X.HCl oxidation for sustainable Cl2recycle over CexZr1-xO2catalysts: effects of Ce/Zr ratio on the activity and stability [J].Industrial & Engineering Chemistry Research, 2014, 53: 19438-19445.
[13]Shan W, Liu F, He H, Shi X Y, Zhang C B.An environmentally- benign CeO2-TiO2catalyst for the selective catalytic reduction of NOxwith NH3in simulated diesel exhaust [J].Catal.Today, 2012, 184(1): 160-165.
[14]Fang J, Bi X, Si D, et al.Spectroscopic studies of interfacial structures of CeO2-TiO2mixed oxides [J].Appl.Surf.Sci., 2007, 253(22): 8952-8961.
[15]Kaneko H, Muiura T, Ishihar H, Taku S, Yokoyama T, Nakajima H, Tamaura Y.Reactive ceramics of CeO2-Mox(M=Mn, Fe, Ni, Cu) for H2generation by two-step water splitting using concentrated solar thermal energy [J].Energy, 2007, 32 :656-663.
[16]Katta L, Sudarsanam P, Thrimurthulu G, Reddy B M.Doped nanosized ceria solid solutions for low temperature soot oxidation: zirconium versus lanthanum promoters [J].Applied Catalysis B: Environmental, 2010, 101(1/2): 101-108.
[17]Liu Chuntao(刘春涛), Shi Pengfei(史鹏飞).CO selective oxidation in hydrogen-rich gas over CuO/CeO2catalysts [J].Journal of Chemical Industry and Engineering(China)(化工学报), 2004, 55(S1): 284-286.
[18]Aramendia M A, Borau V, Jimenez C, Jimenez C, Marinas J M, Porras A, Urbano F J.Synthesis and characterization of ZrO2as an acid-base catalyst dehydration-dehydrogenation of propan-2-ol [J].Journal of the Chemical Society, 1997, 93 (7): 1431-1438.
[19]Bang Y, Seo J G, Youn M H, Song I K.Hydrogen production by steam reforming of liquefied natural gas (LNG) over mesoporous Ni-Al2O3aerogel catalyst prepared by a single-step epoxide-driven sol-gel method [J].International Journal of Hydrogen Energy, 2012, 37(2): 1436-1443.
[20]Jampaiah D, Tur K M, Ippolito S J, Sabri Y M, Tardio J, Bhargava S K, Reddy B M.Structure characterization and catalytic evaluation oftransition and rare earth metal doped ceria-based solid solutions for elemental mercury oxidation [J].RSC Advavaces, 2013, 3: 12963-12974.
[21]Si R, Zhang Y W, Li S J, Lin B X, Yan C H.Urea-based hydrothermally derived homogeneous nanostructured Ce1-xZrxO2(x= 0—0.8) solid solutions: a strong correlation between oxygen storage capacity and lattice strain [J].Journal of Physical Chemistry B, 2004, 108(33): 12481-12488.
[22]Urban S, Tarabanko N, Kanzler C H, Zalewska-Wierzbicka K, Ellinghaus R, Rohrlack S F, Chen L, Klar P J, Smarsly B M, Over H.Stable and active mixed Zr-Ce oxides for catalyzing the gas phase oxidation of HCl [J].Catalysis Letters, 2013, 143(12): 1362-1367.
[23]Xie Xingxing(谢兴星), Fei Zhaoyang(费兆阳), Dai Yong(戴勇), Xu Xihua(徐希化), Chen Xian(陈献), Tang Jihai(汤吉海), Cui Mifen(崔咪芬), Qiao Xu(乔旭).Structure of ceria-based mixed catalytic oxidation performance [J].Journal of Molecular Catalysis(China)(分子催化), 2014, 28(6) :507-514.
