2,3-二取代-5-乙烯基-4,5-二氢呋喃的合成
2015-07-19肖利周志鹏王海飞胡舜钦
肖利,周志鹏,王海飞,胡舜钦
(湖南工业大学包装与材料工程学院,湖南株洲412007)
2,3-二取代-5-乙烯基-4,5-二氢呋喃的合成
肖利,周志鹏,王海飞,胡舜钦
(湖南工业大学包装与材料工程学院,湖南株洲412007)
为解决合成呋喃环需加入较多较昂贵的金属催化剂或者碱的问题,利用-酮酯与对称的烯丙基二碳酸酯为原料,以烯丙基二碳酸酯的物质的量为用量标准,在以占标准用量2%的四(三苯基膦)钯与占标准用量2%的1,2-双- (二苯基膦)乙烷的作用下,合成了一系列的2,3-二取代-5-乙烯基-4,5-二氢呋喃衍生物。试验结果表明,取代基为吸电子基团时反应具有较高的产率,通过改变取代基,已得到了9个不同取代的2,3-二取代-5-乙烯基-4,5-二氢呋喃衍生物,所有产物结构均经1H及13C NMR确证。
-酮酯;烯丙基二碳酸酯;二氢呋喃
0 引言
呋喃(furan)是一种含有4个碳原子和1个氧原子的五元杂环有机物,其结构单元广泛存在于天然产品[1-2]、药品[3]、香料[4]和农用产品[5]等中,此结构单元的存在使得这些物质具有呋喃的结构和功能特性[6],如抗癌、抗艾滋病和抗滤过性病原体等。
在呋喃杂环的制备方法[7-13]中,金属试剂催化合成呋喃环衍生物早已引起人们的注意,选用的金属一般是过渡金属,如铜、金、银和钯等。由于金属催化剂催化合成呋喃环衍生物具有催化剂用量少、反应条件温和及操作简便等优点,促使更多的合成化学家致力于研究发展更多直接有效的合成方法。1994年,Dixneuf小组[12]报道了利用Ru(PPh3) (p-cymene)Cl2催化底物烯炔醇合成呋喃环衍生物的方法,但此方法仅限于含有末端炔基的反应;2004年,Nishizawa小组[13]报道了利用Hg(OTF)2催化合成呋喃环衍生物的研究,此反应虽然反应速度较快,但是体系毒性较大;2007年,Tanimori小组[7]报道了利用[Pd (3-C3H5)Cl]2催化底物烯丙基二碳酸酯与底物-酮酯合成呋喃环的研究,此研究虽能进行反应,但生成物的产率都不高。
C-烯丙基化反应和O-烯丙基化反应是有机化合物分子中碳碳键和碳氧键形成反应中的比较重要的2个反应。本文拟根据烯丙基二碳酸酯与-酮酯通过分子间的C-烯丙基化反应与分子内的O-烯丙基化反应选择性地制备呋喃环衍生物。虽然近年来已有部分文献报道采用此方法[9]合成呋喃衍生物,但其采用较昂贵的催化剂或加入碱才能使反应进行,而本文仅以-酮酯与对称的烯丙基二碳酸酯为原料,以烯丙基二碳酸酯的物质的量为用量标准,以占标准用量2%的四(三苯基膦)钯与占标准用量2%的1,2-双(二苯基膦)乙烷的作用下合成2,3-二取代-5-乙烯基-4,5-二氢呋喃,此反应具有催化剂用量少、反应条件温和及操作简便等优点。本文拟通过筛选不同配体与溶剂种类以及金属催化剂与配体的用量为研究内容,研究它们对合成呋喃环衍生物的影响,确定最佳的反应条件和可能的反应机理,并通过1H及13C核磁共振(nuclear magnetic resonance,NMR)对合成的产物进行确证,以期获得不同取代基的呋喃环衍生物。
1 试验部分
1.1 试剂与仪器
石油醚、乙酸乙酯(ethyl acetate,AcOEt)、二氯甲烷(dichloromethane,DCM),均为分析纯,天津津东天正精细化学试剂厂生产;甲苯(methylbenzene,Toluene)、四氢呋喃(terahydrofuran,THF)、1,2-二氯乙烷(1,2-dichloroethane,EDC)、甲醇(methyl alcohol,MeOH)、1,4-二氧六环、吡啶、碳酸二甲酯、氯甲酸乙酯、顺式1,4-丁烯二醇,均为分析纯,上海泰坦科技股份有限公司生产;氢化钠,质量分数为60%,萨恩化学技术(上海)有限公司生产;1,2-双(二苯基膦)乙烷(1,2-bis(diphenylphosphino)ethane,DPPE)、三苯基膦(triphenyl phosphine,PPh3)、1,1’-双(二苯基膦)二茂铁(1,1’-bis(diphenylphosphino)ferroce,DPPF)、1,4-双(二苯基膦)丁烷(1,4-bis(diphenyphosphino)butane,DP P B)、1,3-双(二苯基膦)丙烷(1,3-bis (diphenylphosphino) propane,DPPP)、四(三苯基膦)钯((beta-4)-platinum,Pd(PPh3)4),均为分析纯,萨恩化学技术(上海)有限公司生产;苯乙酮、对甲氧基苯乙酮、间甲氧基苯乙酮、2-萘乙酮、对氟苯乙酮、邻氟苯乙酮、邻溴苯乙酮、对溴苯乙酮、间溴苯乙酮,均为分析纯,阿拉丁试剂(上海)有限公司生产。
核磁共振仪,Bruker AV-400 MHz型,瑞士Bruker公司生产;数显智能控温磁力搅拌器,SZCL-3B型,巩义市予华仪器责任有限公司生产;分析电子天平,JA1003型,上海舜宇恒平科学仪器有限公司生产;三用紫外分析仪,ZF-6型,上海嘉鹏科技有限公司生产;远红外快速恒温干燥箱,YHG-600S型,上海跃进医疗器械厂生产;旋转蒸发仪,R201D型,郑州市亚荣仪器有限公司生产。
1.2 试验过程
本研究按照图1所示的实验流程图制备呋喃环衍生物。首先,根据文献合成-酮酯1a~1i与对称的烯丙基二碳酸酯2,再将合成的-酮酯1a~1i与烯丙基二碳酸酯2在一定条件下反应合成相应的呋喃环衍生物3a~3i。

图1 呋喃衍生物的合成流程图Fig. 1The flowchart for furan derivatives synthesis
0℃条件下,在50 mL烧瓶中加入氢化钠15 mmol与甲苯10 mL,搅拌10 min,再将含有相应取代基的苯乙酮5 mmol与甲苯2.5 mL的混合液逐滴加入烧瓶内,回流1 h;待温度降至0℃后,将碳酸二甲酯10 mmol逐滴加入其中,滴加完全后回流3~6 h;采用薄层色谱法(thin-layer chromatography,TLC)点板跟踪检测至原料点消失,冷却后滴加冰醋酸至糊状物不再增加,再向其中加入冰水至糊状物完全溶解。采用乙酸乙酯提取,并用无水硫酸镁干燥,过滤,浓缩,柱层析纯化得相应的-酮酯1a~1i。[14-16]
2)烯丙基二碳酸酯2的制备
在0℃条件下,将顺式1,4-丁烯二醇10 mmol与吡啶2.5 mmol加入到含有33 mL二氯甲烷溶剂的烧瓶中,搅拌均匀,再将氯甲酸乙酯25 mmol逐滴加入其中;在0℃条件下搅拌10 min,再在室温下搅拌1 h,采用薄层色谱法点板跟踪检测至原料点消失,加入二氯甲烷33 mL,用浓度为1 mol/L稀盐酸33 mL进行3次萃取,萃取后的有机相用饱和碳酸钠溶液33 mL进行3次洗涤,再用饱和氯化钠溶液33 mL进行3次洗涤至中性;干燥,浓缩,柱层析纯化得无色液体产物。[17-18]
3)呋喃环衍生物的合成
准确量取四(三苯基膦)钯0.01 mmol、1,2-双(二苯基膦)乙烷0.01 mmol与-酮酯1a~1(i0.75 mmol)加入到含有1.5 mL二氯甲烷溶剂的烧瓶中,搅拌均匀,再将0.5 mmol烯丙基二碳酸酯2加入烧瓶中,室温搅拌4~6 h,采用薄层色谱法点板跟踪检测至原料点消失,反应体系不需其它操作,直接柱层析纯化得油状液体产物3a~3i。
1.3 产物的表征与检测
合成的呋喃环衍生物经1H及13C NMR确认,其部分产物的核磁谱图如图2所示。
以2-苯基-3-羧基甲酯-5-乙烯基-4,5-二氢呋喃的核磁谱图为例,从图2a中13C NMR可看出,化学位移在100以下的归属于饱和基团上的碳,而化学位移在100以上的归属于不饱和基团上的碳,由13C NMR可以粗略地推算出碳原子的个数。再对图2b中的1H NMR进行分析,可进一步确认,其化学位移在7.73~7.28之间的归属于苯环上的氢,化学位移在5.98~5.05之间的归属于烯基上的氢,化学位移在3.25~2.81之间的归属于亚甲基上的氢,化学位移为3.59的单峰归属于甲氧基上的氢。其他物质的谱图,例如2-(4-甲氧基-苯基)-3-羧基甲酯-5-乙烯基-4,5-二氢呋喃(图2c,2d)中的给电子取代基与2-间溴苯基-3-羧基甲酯-5-乙烯基-4,5-二氢呋喃(图2e,2f)中的吸电子取代基,都可采用类似的方法进行确证。含有吸电子基团或给电子基团,其化学位移会有所变化,但不大,从氢谱和碳谱上看,含有给电子基团使部分氢和碳的化学位移向高场移动,而吸电子基团使其化学位移向低场移动。

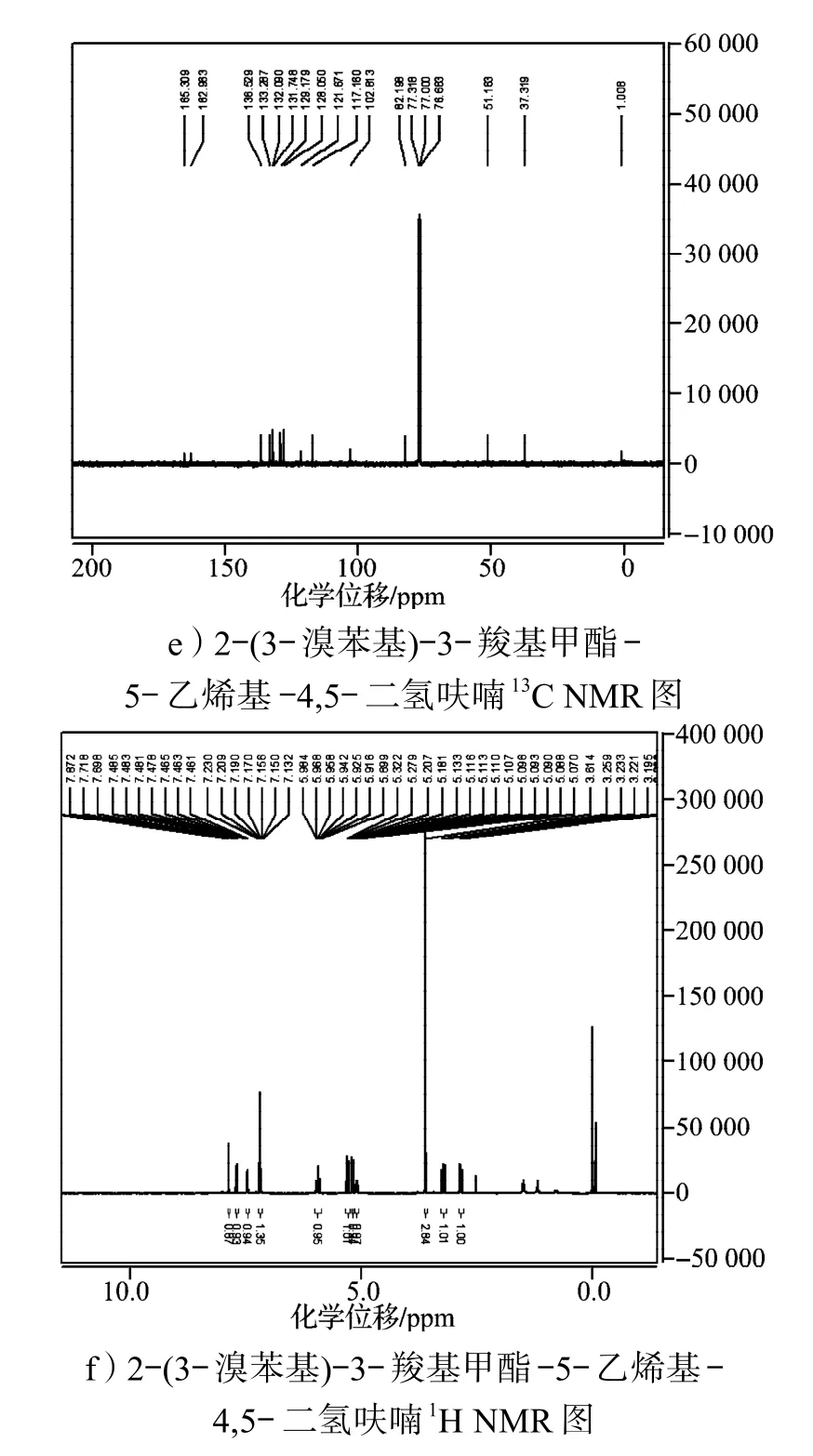
图2 部分呋喃环衍生物的核磁共振谱图Fig.2 The NMR spectra of part of furan derivatives
2 结果与讨论
2.1 试验条件的优化
以0.75 mmol 苯甲酰乙酸甲酯1a与0.5 mmol 烯丙基二碳酸酯2为反应底物来考察它们在四(三苯基膦)钯(Pd(PPh3)4)催化条件下得到的呋喃环衍生物。以图3所示的反应对试验条件进行优化。
图3 2-苯基-3-羧基甲酯-5-乙烯基-4,5-二氢呋喃的合成Fig.3 The synthesis of 2-phenyl-3-carboxylate-methyl-5-vinyl-4,5-dihydrofuran
为确证其它因素(如配体、溶剂等)对试验结果的影响,现对其进行筛选(如表1)。首先是配体种类的筛选,如1,2-双(二苯基膦)乙烷(DPPE)、1,1’-双(二苯基膦)二茂铁(DPPF)、1,4-双(二苯基膦)丁烷(DPPB)、1,3-双(二苯基膦)丙烷(DPPP)和三苯基膦(PPh3);其次是溶剂种类的筛选,如THF,MeOH,EDC,Toluene,1,4-Dioxane和DCM;最后是金属催化剂和配体的用量确定等。

表1 合成条件的优化Table 1Optimization of synthetic conditions
通过观察金属催化剂与配体作用时发现(表1,序号1~2),Pd(PPh3)4单独作用下不能催化合成呋喃环,只有在与配体共同作用时才能使此反应进行,生成呋喃衍生物。配体筛选时发现(表1,序号1,3~6),DPPE较其他配体作用时有较好的反应活性和产率,其他配体如DPPF,DPPB,DPPP和PPh3与金属催化剂共同作用下虽都能促进反应进行,生成呋喃环,但其反应速度都较DPPE作用时慢,生成物的产率也较低。在其它条件不变的条件下,以DPPE为配体进行溶剂筛选时发现(表1,序号1,7~11),分别以DCM和EDC作为反应溶剂时,反应情况较好,反应体系较纯净,以THF,MeOH,Toluene、1,4-Dioxane作为反应溶剂时,虽有产物生成,但是产率却不是很高,尤以1,4-Dioxane作为溶剂时,反应体系较复杂且产率极低(小于5%)。但是以DCM与EDC作为反应溶剂时,DCM较EDC有较好的反应活性与反应速率,且DCM作为溶剂时只需用4A分子筛简单除水即可,不需经过严格的去水处理。总的来说,不含氧溶剂(如DCM,EDC,Toluene)的反应速率较含氧溶剂(如MeOH,1,4-Dioxane)快。确定反应的加入量进行时发现(表1,序号11~14):以烯丙基二碳酸酯的物质的量为用量标准,当金属催化剂Pd(PPh3)4用量为标准用量的2%、配体DPPE用量为标准用量的2%时,达到最优用量,其它比例的用量造成原料的浪费或使反应不彻底。对各物质种类及其用量进行确定后,再对条件温度进行优化。将温度条件分别设为:较低温度-15℃与0℃;较温和温度,即10℃与25℃;二氯甲烷沸点温度,40℃。试验结果发现,在-15 ℃及0℃条件下,反应速率慢于其他3个温度条件下的反应速率,且生成物产率较低,而在10℃,25℃与40 ℃温度条件下的反应速率相差不大,且各生成物产率也相差不大,故将反应温度条件设为室温。
通过上述对各条件的筛选,确定此反应的最优化条件如下:底物-酮酯1a为0.75 mmol、底物烯丙基二碳酸酯2为0.5 mmol、催化剂四(三苯基膦)钯为0.01 mmol、配体1,2-双(二苯基膦)乙烷为0.01 mmol、溶剂二氯甲烷为1.5 mL、室温。
2.2 系列呋喃衍生物的合成
图4 系列呋喃衍生物的合成Fig.4 The synthesis of a series of furan derivatives
通过对表2分析可知:苯环上取代基的种类对反应产率有较大的影响,当苯环上增加取代基后,其反应产率有所增加。为了观察电子效应对反应结果的影响,本课题组研究了苯环上含有给电子基团的甲氧基取代基及吸电子基团的氟与氯取代基,观察的结果表明,电子效应对反应的影响较大,吸电子基团较给电子基团有较高的产率,尤其是含有间溴取代基的,其产率达到83%。分析苯环上取代基位置对反应的影响时发现:不论是吸电子基团取代基(序号5~6与序号7~9)还是给电子基团取代基(序号2~3),苯环上同一位点不同的取代基(序号2,5,9;序号3,8与序号6~7)对试验结果有较大影响,同一取代基(序号2~3;序号5~6与序号7~9)不同的位点对试验结果亦有较大的影响。从表2可看出,不论是给电子基团还是吸电子基团,反应速率都较快,能在2.5~6.0 h内反应完全,并达到较高的产率。

表2 系列呋喃衍生物的合成Table 2The synthesis of a series of furan derivatives
2.3 反应机理的探讨
依据文献[19]类似机理的报导与对表1和2的试验结果分析,提出了下面的可能机理:首先烯丙基二碳酸酯在零价钯作用下脱除单碳酸酯负离子生成配合物I,紧接着单碳酸酯负离子进攻-酮酯生成互变异构负离子IIA和IIB,然后碳负离子IIA通过分子间的C-烯丙基化反应进攻配合物Ⅰ生成中间体III,中间体III继续通过和零价钯配位、脱碳酸酯,并通过分子内的O-烯丙基化反应选择性地得到呋喃环衍生物3a。其可能的反应机理如图5所示。

图5 反应机理的探讨流程图Fig.5 The flowchart for the reaction mechanism investigation
3 结语
由于含呋喃骨架的化合物广泛存在于天然产物中,并具有多种生物活性,如:抗癌、抗菌等,此类衍生物的合成引起众多从事有机合成者的关注。利用-酮酯与对称的烯丙基二碳酸酯为原料,以烯丙基二碳酸酯的物质的量为用量标准,在以占标准用量2%的四(三苯基膦)钯与占标准用量2%的1,2-双-(二苯基膦)乙烷的作用下,通过分子间的C-烯丙基化反应与分子内的O-烯丙基化反应在一定条件下以中等的产率得到一系列的含有呋喃骨架的2,3-二取代-5-乙烯基-4,5-二氢呋喃衍生物。试验结果表明:电子效应对实验结果有较大的影响,吸电子基团较给电子基团有较高的活性及产率;不同的取代基对试验结果有影响,取代基的位置对反应结果的影响也较大。此方法具有催化用量少、反应条件温和及操作简便等优点,该研究为后续正在进行的的手性呋喃环衍生物的合成提供了依据,也为呋喃类药物的合成与研究提供了新的思路与方法。
[1]Rahmathullah S M,Hall J E,Bender B C,et al. Prodrugs for Amidines:Synthesis and Anti-Pneumocystis Carinii Activity of Carbamates of 2,5-Bis(4-Amidinophenyl)Furan [J]. Journal of Medicinal Chemistry,1999,42(19):3994-4000.
[2]Florio R D,Rizzacasa M A. Synthesis of 2,2-Disubstituted Furanoid Natural Products:Total Synthesis of Sphydrofuran[J]. Journal of Organic Chemistry,1998,63 (23):8595-8598.
[3]Anbazanagn M,Boykin D W. A Facile Synthesis of the Prodrug 2,5-Bis(4-0-Methoxyamidinophenyl)Furan and Analogs[J]. Heterocyclic Communications,2003,9(2):117-118.
[4]Kraft P,Baigrowicz J A,Denis C,et al. Odds and Trends:Recent Developments in the Chemistry of Odorants[J]. Angewandte Chemie International Edition,2000,39(17):2980-3010.
[5]Nizamuddin,Madhuri G,Kumar S M. Synthesis and Fungicidal Activity of Some Furan Containing Azoles[J]. Journal of Scientific & Industrial Research,1999,58(12):1017-1020.
[6]Sugimura H,Osumi K,Kodaka Y,et al. Stereoselective Synthesis of 2’-Deoxy--D-Threopentofurano Nucleosides by the NBS-Promoted Coupling Reaction of Thioglycosides with Silylated Heterocyclic Bases[J]. Journal of Organic Chemistry,1994,59(25):7653-7660.
[7]Tanimori S,Kirihata M. Palladium-Catalyzed Approach to Substituted Dihydrofurans,Cyclopenta[b]Furans and Tetrahydrobenzofurans[J]. Synthesis,2007(1):39-44.
[8]Bowman R K,Johnson J S. Nickel-Catalyzed Rearrangement of 1-Acyl-2-Vinylcyclopropanes:A Mild Synthesis of Substituted Dihydrofurans[J]. Organic Letters,2006,8(4):573-576.
[9]Tanimori S,Kato Y,Kirihata M. Simple Preparation of New Functionalized Furan Derivatives Via Sequential C-C and C-O Bond Formation Mediated by Palladium-Phosphine Catalyst[J]. Synthesis,2006(5):865-869.
[10]Ohno A,Yamane M,Hayashi T,et al. Preparation and Use of Chiral Ferrocenylphosphines Containing New Alkyl Substituents on the Ferrocenylmethyl Position[J]. Tetrahedron,1995,6(10):2495-2502.
[11]Murakami H,Matsui Y,Ozawa F,et al. Cyclodehydration of Cis 2-Butene-1,4-Diol with ActiveMethylene Compounds Catalyzed by a Diphosphinidenecyclobutene-Coordinated Palladium Complex[J]. Journal of Organometallic Chemistry,2006,691(4):3151-3156.
[12]Sieller B,Bruneau C,Dixneuf P H. Novel Ruthenium-Catalysed Synthesis of Furan Derivatives Via Intramolecular Cyclization of Hydroxy Enynes[J]. Chemical Communications,1994(4):493-494.
[13]Hashmi A S K,Sinha P. Gold Catalysis:Mild Condition for the Transformaion of Alkynyl Epoxides to Furans[J]. Adv. Synth. Catal.,2004,346(4),432-438.
[14]Stergiou A,Bariotaki A,Kalaitzakis D,et al. Oxone-Mediated Oxidative Cleavage of-Keto Esters and 1,3-Diketones to -Keto Esters and 1,2-Diketones in Aqueous Medium[J]. The Journal of Organic Chemistry,2013,78 (14):7268-7273.
[15]Liu X,Cheng R,Zhao F F,et al. Direct-Acyloxylation of Enamines Via PhIO-Mediated Intermolecular Oxidative C-O Bond Formation and Its Application to the Synthesis of Oxazoles[J]. Organic Letters,2012,14(21):5480-5483.
[16]Russo A,Galdi G,Croce G,et al. Highly Enantioselective Epoxidation Catalyzed by Cinchona Thioureas:Synthesis of Functionalized Terminal Epoxides Bearing a Quaternary Stereogenic Center[J]. Chemistry a European Journal,2012,18(20):6152-6157.
[17]Morgen M,Bretzke S,Li P F,et al. Stereodivergent Synthesis of 1,3-Syn-and-Anti-Tetra-Hydropyrimidinone-S[J]. Organic Letters,2010,12(20):4494-4497.
[18]Hodgson D M,Stefane B,Miles T J,et al. Organolithium-Induced Alkylative Ring Opening of Aziridines:Synthesis of Unsaturated Amino Alcohols and Ethers[J]. The Journal of Organic Chemistry,2006,71(22):8510-8515.
[19]Hu Z J,Li Y G,Liu K,et al. Bis(Perfluoroalkyl) Phosphino-Oxazoline:A Modular,Stable,Strongly -Accepting Ligand for Asymmetric Catalysis[J]. The Journal of Organic Chemistry,2012,77(18):7957-7967.
(责任编辑:申剑)
The Synthesis of 2,3-Bisubsituted-5-Vinyl-4,5-Dihydrofuran
Xiao Li,Zhou Zhipeng,Wang Haifei,Hu Shunqin
(School of Packaging and Materials Engineering,Hunan University of Technology,Zhuzhou Hunan 412007,China)
In order to solve the problems of adding expensive metal catalyst or alkali to synthesize furan ring,taking symmetrical allyl carbonate as the dosage standard,applies-ketoester and symmetrical allyl carbonate as raw materials to synthesize 2,3-bisubsituted-5-vinyl-4,5-dihydrofuran derivatives under the combined catalysis of the standard 2% tetrakis (triphenylphosphine)palladium catalyst and of the standard 2% 1,2-bis(diphenylphosphino)ethane. The experimental results show that the reaction has high yields with the electron withdrawing groups. By changing the substituents,obtains 9 different substituted 2,3-bisubsituted-5-vinyl-4,5-dihydrofuran derivatives. All products are confirmed by1H and13C NMR.
-ketoester;allyl carbonate;dihydrofuran
O626.11
A
1673-9833(2015)01-0102-07
2014-09-03
国家自然科学青年基金资助项目(21202042),湖南省自然科学青年基金资助项目(13JJ4090)
肖利(1989-),男,湖南洞口人,湖南工业大学硕士生,主要研究方向为有机催化合成,E-mail:5939702712@qq.com
胡舜钦(1967-),男,湖南桃江人,湖南工业大学教授,博士,主要从事分析化学方面的研究,E-mail:1328235513@qq.com
10.3969/j.issn.1673-9833.2015.01.019
