气相色谱法测定采后芒果中苯醚甲环唑残留量的测量不确定度评定
2015-07-10方宗壮张小玲潘晓威赵振东谢艳丽章程辉
方宗壮,张小玲,潘晓威,赵振东,谢艳丽,章程辉
(1. 海南大学 食品学院,海南 海口570228;2. 海南大学分析测试中心,海南 海口570228)
苯醚甲环唑属三唑类杀菌剂,由于其对蔬菜和瓜果等多种真菌性病害具有非常好的保护和治疗作用,因此,近年来它在果树、蔬菜、小麦、豆类、瓜类等作物中被广泛使用. 苯醚甲环唑为低毒类物质,将其直接喷洒于蔬菜、水果上,人长期低剂量的反复接触可能会有蓄积作用而使人们易受其危害[1].
目前,对食品中的苯醚甲环唑的检测已多见于报道,对其检测方法和检测结果的要求也越来越高,故采取不确定度分析的方法,分析其测量过程中影响测量结果准确性的因素并对其改进是非常必要的[2-7].
本文通过对采后芒果中苯醚甲环唑残留量整个测定过程的监控,运用不确定度分析的方法,找出了其测定过程中影响结果准确性的主要因素.
不确定度是指因系统影响或人为测量误差的存在而导致被测量值不能确定的程度. 不确定度越小,被测值的测量结果与其真值就越接近;反之,测量结果与真实值的差别就越大.不确定度一方面可用于判断被测值是否准确;另一方面可用于测量结果准确性之间的比较.本文采用测量不确定度来表示指南中的办法确定的不确定度,其一般流程如下:
分析并确定不确定度来源 根据不确定度来源的性质确定评定类别(A 或B 类) 按所属类别评定方法评定标准不确定度 计算合成标准不确定度 确定扩展不确定度
1 材料和方法
1.1 材料与试剂 芒果采自海南省昌江县芒果基地,品种为台农,药品标样为苯醚甲环唑标准品(纯度大于98.5%),由农业部质量标准研究所提供;实验农药为w =10%的苯醚甲环唑水分散粒剂,由浙江世佳科技有限公司提供.
乙腈(分析纯),广东光华化学厂有限公司;乙腈(色谱纯),Buridick & Jackson;无水硫酸镁(分析纯),广州化学试剂厂;氯化钠(分析纯),广州化学试剂厂;PSA(乙二胺-N -丙基硅烷)孔径6 nm,粒度45 μm,爱杰尔公司.
1.2 实验仪器 安捷伦6890N 型电子俘获气相色谱仪,美国安捷伦公司;IKA MS3 涡旋器,德国IKA 公司;RV 10 旋转蒸发仪,申生科技公司;H1650 高速离心机,湘仪离心机公司;食品料理机;AA.160 型电子天平,美国丹法公司;HS-840/HS-1300 超净工作台,苏州尚田洁净公司;高速匀浆分散机,德国IKA 公司;BD/BC-199VMQ 冰柜,美的电器.
1.3 苯醚甲环唑标准溶液的配制 标准母液:以万分之一电子分析天平称取0.010 00g 苯醚甲环唑标准品,用丙酮定容至10 mL,得到1 000.0 μg·mL-1的标准母液.保存于-18 ℃冰箱中,用时稀释至相应质量浓度.
1.4 实验仪器参数 色谱柱:安捷伦DB-1,30 m×0.25 mm ×0.25 μm;进样口温度:200 ℃;检测器温度:350 ℃;柱温:210 ℃保持1 min,然后以10 ℃·min-1升至280 ℃保持8 min;分流比10∶1.
1.5 测定方法及步骤 将芒果样的可食部分用刀切下,用九阳食物料理机搅碎、混匀,作为备用.然后切取不少于200 g 芒果全果,充分混匀高速匀浆2 min,制成待测试样.
(1)提取:准确称取5 g(准确到0.01 g)待测芒果样品
取50 mL 的离心管,向其中加入10 mL 乙腈,混匀后再加入4 g 无水MgSO4和1 g NaCl,再混匀,于6 000 r·min-1离心5 min.
(2)净化:取离心后上清液5 mL 添加到10 mL 离心管中,再加入100 mg PSA、500 mg 无水MgSO4,涡旋混匀1 min 后,6 000 r·min-1离心10 min,吸取4 mL 上清液,旋蒸至近干,加入2 mL 正己烷溶解,0.22 μm 滤膜过滤,待测.
1.6 数学模型
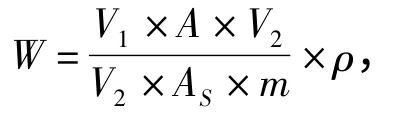
式中:W—试验中被测农药的残留量(mg·kg-1);ρ—标准溶液中农药的质量浓度(mg·L-1);A—样品溶液中目标物的峰面积;AS—农药标准溶液中目标物的峰面积;V1—提取溶剂的总体积(mL);V2—吸取出提取液的体积(mL);V3—样品最后定容的体积(mL);m—试样的质量(g).
1.7 测量不确定度的来源分析 从检测过程来分析,导致气相色谱法测定采后芒果中苯醚甲环唑残留量的不准确因素主要有标品浓度、样品及标品的峰面积、样品的处理方法以及称量、测量、定容等方面[8].
2 结果与分析
2.1 标液的质量浓度ρ 引起的标准不确定度u(ρ) 标液的质量浓度标准不确定度u(ρ)来源于标准品的称量,在标准品溶液的配制过程,对各不确定度分量需分别进行评定,然后合成.
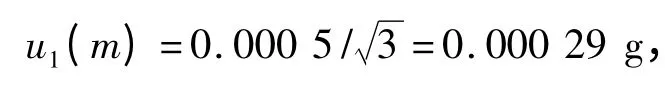
重复性误差为± 0.000 1 g,则:
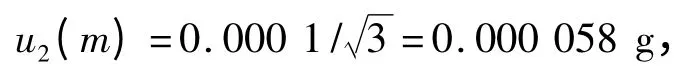
则天平称量是造成的标准不确定度为:
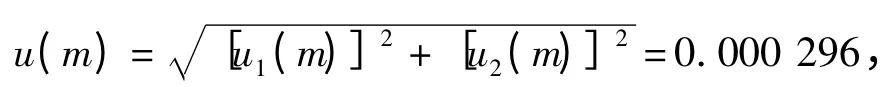
相对不确定度:urel(m)=u(m)/m=0.029 6.
2.1.2 标品溶液配制过程造成的标准不确定度u(V )
2.1.2.1 标品溶液配制过程中量器校准造成的标准不确定度u1(V)标品溶液配制过程中使用了一系列的玻璃量器,根据检定规程[9],均有相应的最大允许误差,取计算各玻璃量器的标准不确定度(表1).
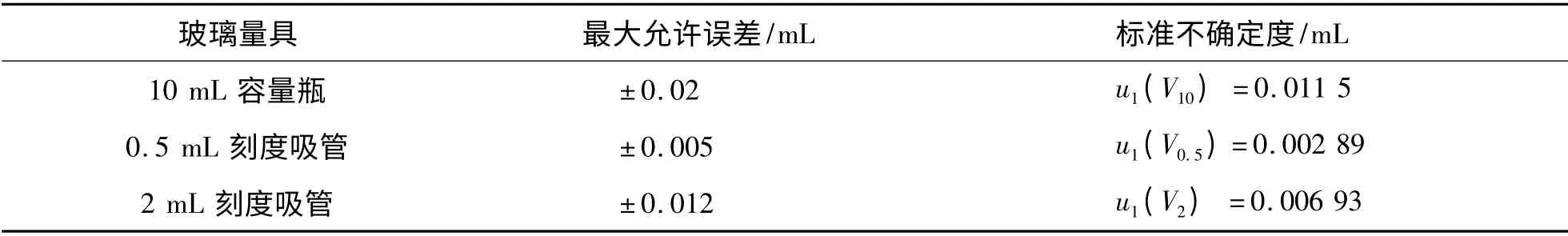
表1 量器校准造成的标准不确定度
2.1.2.2 标品溶液配制过程中因人读数造成的标准不确定度u2(V)依据《化学分析中不确定度评估指南》,人员读数允许有1% 的不确定性,即±0.01 的误差,取计算各标准不确定度(表2).

表2 人员读数造成的标准不确定度
2.1.2.3 标品溶液配制过程中造成的相对标准不确定度urel(V)(表3).

表3 定容稀释造成的相对标准不确定度
2.1.3 标品浓度ρ 造成的相对标准不确定度[urel(ρ)]
庭园内主要的建筑物都安排在同一个轴网系统内,布置了方正的建筑之后,剩下不规则形状的用地则通过安排石山、树池、花池等配景去消解。树池和花池的平面形状在靠近建筑的一侧,保持与建筑外墙平行或垂直。在不规则形状的用地里,走道与花池使用了不规则的折线造型,修饰了不规则的用地形状(图16)。
由于urel(m),uRel(V1),uRel(V2),uRel(V3)是互相独立和互不相关的,故标品的浓度ρ 造成的相对标准不确定度:

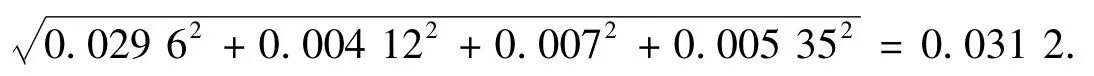
2.2 样品的峰面积A1造成的标准不确定度u(A1) 样品峰面积的标准不确定度u(A)来源于气相色谱仪的定量重复性.依据气相色谱仪的检定证书,其定量重复性小于2.0%,取得其相对不确定度为:

2.3 标准品的峰面积A2造成的标准不确定度u(As) 同2.2 法的分析,得标准品峰面积的标准不确定度urel(As)=0.011 54.
2.4 样品处理过程中体积V 造成的标准不确定度V(v)和相对标准不确定度Vrel(v)
2.4.1 因量器校准造成的标准不确定度V1(v) 样品处理过程中使用了一系列量器,均有最大允许误差,取计算标准不确定度(表4).
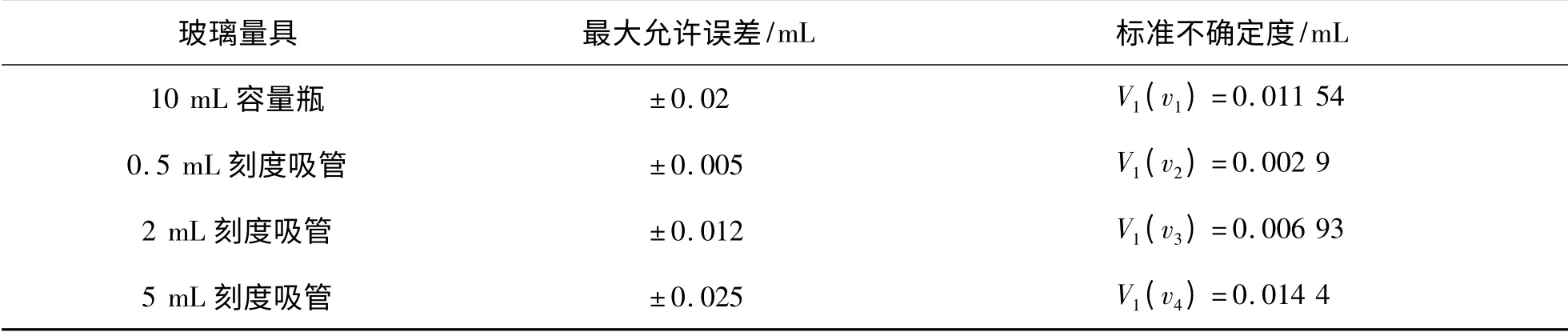
表4 样品处理过程中量器校准造成的标准不确定度
2.4.2 人员读数造成的标准不确定度V2(v) 同2.1.2.2 方法的分析,得标准不确定度(表5).

表5 人员读数造成的标准不确定度
2.4.3 样品处理过程中体积V 造成的相对标准不确定度 由表6 可得:

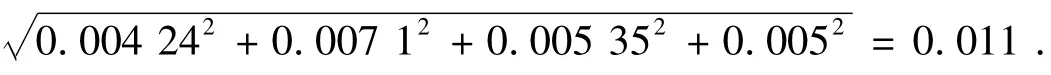

表6 体积V 造成的相对标准不确定度
2.5 样品称样量m 造成的标准不确定度u(m) 称样品5.00 g,电子天平检定的最大允许误差为±0.000 5 g ,故得:

重复性的误差为± 0.000 1 g,得
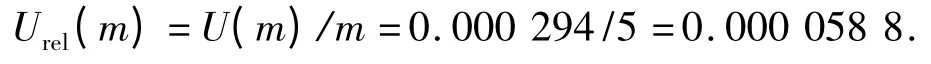
则可得天平校准的标准不确定度和相对标准不确定度分别为:
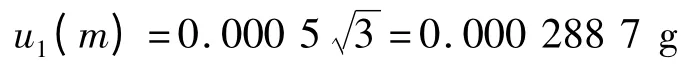

2.6 样品测量重复性造成的标准不确定度u(x) 称3 份样品,分别测定,测定结果如表7 .用极差法计算得:
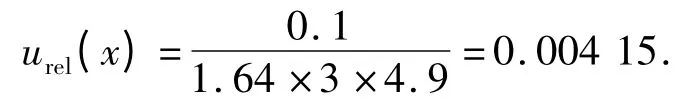

表7 样品重复性测量结果
2.7 样品中加标回收率造成的标准不确定度u(R) 样品的处理过程是非常复杂的,容易导致测量误差,故采用空白加标测定的办法.在回收率实验中做3 份空白加标,分别测定,用极差法计算得:

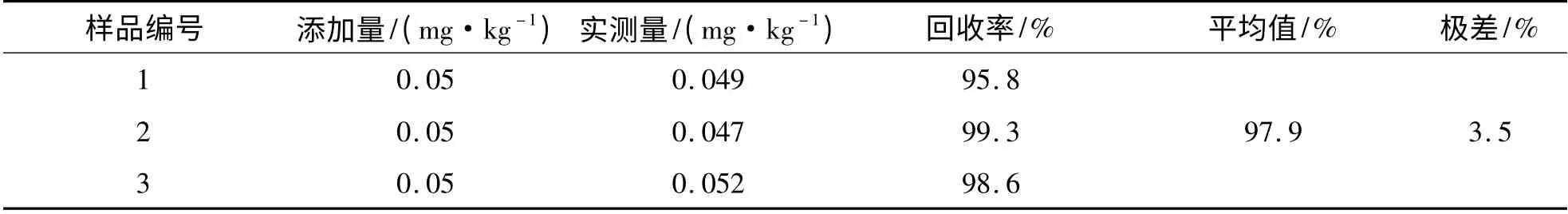
表8 空白加标回收率测定结果
2.8 合成标准不确定度及扩展不确定度 相对合成标准不确定度为:


实验所得采后芒果中苯醚甲环唑残留量为0.002 5 mg·kg-1,则测量结果的合成标准不确定度为:u(x)=0.002 5 mg·kg-1× 0.037 8 =0.000 094 5 mg·kg-1;取k=2,则测量扩展不确定度为:U=2u(x)=2 ×0.000 094 5 mg·kg-1=0.000 189 mg·kg-1.
2.9 测量结果表示 采后芒果中苯醚甲环唑残留量可表示为(0.002 5 ±0.000 189)mg·kg-1,k=2.
3 讨 论
由于样品的处理和配制过程严格按照实验室的标准操作规程,并且实验中所使用的仪器及玻璃器具皆属高精密度用品,故样品处理和配置过程中因转移、稀释、定容和因仪器精密度造成的不确定度可忽略不计.但整个评定过程中造成不确定度大的主要因素有标品浓度以及样品和标品的峰面积,分析其原因,主要是由于厂家标品纯度不高或标品配置后储藏过久导致纯度下降所致,还有气象色谱仪柱效下降的问题,故可选用标品应挑选质量、纯度更好的厂家,标品要现配现用.
4 结 论
通过对气相色谱法测定采后芒果中苯醚甲环唑残留量过程的监测,得出影响其测量结果准确性的因素主要是标品的浓度,样品和标品的峰面积,故测定采后芒果中苯醚甲环唑残留量的过程中,应对其足够重视.
该方法同样适用于气相色谱法测定蔬菜、水果中其他农药残留的测量不确定度评定,选择恰当的实验方法,规范操作,完全可以降低测量的不确定度,保证实验结果的准确、可靠性.
[1]王筱芬,谢琳,史岩.苯醚甲环唑的毒性及致突变性试验研究[J]. 职业与健康,2005,21(12):1 953 -1 954.
[2]崔淑华,郭庆龙,刘冰,等.液相色谱法测定鸡肉中二氯二甲吡啶酚残留量的测量不确定度评定[J].食品科学,2007,28(12):397 -400.
[3]卢丽明,黄诚.HPLC 法测定碳酸饮料中日落黄含量的不确定度分析[J].中国热带医学,2008,8(8):1 448 -1 449.
[4]张素娟.气象色谱法测定白酒中乙酸乙酯含量的不确定度分析[J].食品科学,2010,31(2):151 -152.
[5]周艳明,汪霞.气相色谱法测定西红柿中乙烯利残留量的不确定度分析[J].食品工业科技,2008,29(1):278 -280.
[6]高素虹.重量法测定食品中灰分的测定不确定度分析[J]. 海峡预防医学杂志,2006,12(5):39 -40.
[7]谢显传,王冬生.果蔬类农产品中农药残留量检测的不确定度评定[J].化学分析计量,2006,15(2):7 -9.
[8]国家质量监督检验检疫总局.JJF1135—2005 化学分析测量不确定度评定[S].北京:中国计量出版社,2005.
[9]国家质量监督检验检疫总局.JJG196—2006 常用玻璃量器[S].北京:中国计量出版社,2007.
[10]林小莉,董艳峰,于静泉. 高效液相色谱法测定鸡肉中噁喹酸残留量的测量不确定度评定[J]. 食品科学2009,30(14):219.
[11]柯瑞华.化学成分测量不确定度的评定.冶金分析[J].2004,24(1):63 -72.
[12]郑怀东,刘学光,关丽,等. 气相色谱法测定河蟹中多氯联苯残留量的不确定度分析[J].食品科学.2011,32(16):334.
