MgH分子特性和势能随外电场的变化规律
2015-07-07伍冬兰温玉峰万慧军谢安东
伍冬兰,谭 彬,温玉峰,万慧军,谢安东
(井冈山大学数理学院,江西吉安 343009)
MgH分子特性和势能随外电场的变化规律
伍冬兰,谭 彬,温玉峰,万慧军,谢安东
(井冈山大学数理学院,江西吉安 343009)
采用密度泛函B3P86方法,结合Dunning的相关一致五重基cc-PV5Z,设置不同外电场参数进行优化计算,获得MgH分子在不同外电场中的键长、偶极矩、振动频率和红外光谱强度等物理性质参数。在此基础上,采用能量高精度耦合簇CCSD(T)方法和相同的基组扫描计算单点能,获得不同外电场下的势能曲线。结果分析表明,物理性质参数和势能均随外电场的变化而变化,且外加反向电场时变化幅度更明显。考虑到外电场与分子的相互作用,本文引入偶极近似构建外电场中的势能函数模型,编制程序拟合对应的势能函数,得出拟合参数,进而计算临界离解电场参数,结果与数值计算和理论分析较一致,相对误差均在3%以内,说明构建的模型是合理可靠的。这为分析外电场中分子光谱、动力学特性和分子Stark效应冷却囚禁提供重要的理论和实验参考。
MgH分子;势能函数模型;外电场
镁及镁氢化物具有储氢量高、质量轻、成本低、无污染、资源丰富和便于运输等综合优势,有望作为最有发展前景的氢能载体材料而备受重视[1-3]。双原子氢化物MgH是元素周期表中较简单的极性自由基分子,具有较大的固有电偶极矩,在外电场中会受电场偶极力作用,是分子交、直流Stark效应冷却的重要候选分子[4]。通过实验测量和理论计算光谱性质是目前研究分子结构常用的方法和手段,已采用不同的方法对MgH自由基分子进行了广泛的实验和理论研究。1986年,Leopold等[5]测量了MgH分子的远红外光谱;1988年,Lemoine等[6]测量了MgH和MgD分子的红外激光光谱;1990年,Zink等[7]在实验室测量并指认了天体物理中的 MgH分子。理论研究方面,Mostafanejad等[8]分析了MgH分子的X2Σ+、А2Π和B2Σ+的势能函数和跃迁偶极矩;Mestdagh等[9]采用ic-MRCI方法研究了MgH分子基态和激发态的势能函数和分子常数,同时得到了较精确的光谱常数;宇燕[10]采用CCSD(T)方法研究了MgH分子基态结构和势能函数,得出其光谱数据;吕兵等[11]采用QCISD(T)方法分析了MgH自由基分子的基态和低激发态的势能函数及光谱常数;张萍等[12]研究了MgH自由基分子及离子的结构和性质,得到其光谱常数和垂直电离势。从这些结果可看出,实验和理论计算大多集中于分析MgH自由基分子势能函数和光谱常数,对外电场作用下分子结构、势能函数和光谱的研究较少,而在分子冷却和激光场实验等相关研究领域中通常需外加电场,这会增加外电场与分子体系的相互作用,从而导致分子的结构、物理特性及势能函数等发生变化[13-15]。因此,有必要对外电场中的分子精确结构、物理性质、势能函数与光谱等进行更细致分析,为进一步进行分子冷却实验提供重要的参考。
本文采用B3P86和cc-PV5Z方法优化计算不同外电场中MgH分子的结构和物理性质,分析外电场对其几何构型和物理性质参数的影响,判断离解电场所处范围;设置合适的电场参数,采用能量高精度耦合簇CCSD(T)方法,扫描计算该范围的单点能,获得其势能曲线,再利用构建的势能函数模型编制程序拟合势能曲线,得出拟合参数,与数值计算和理论分析进行比较,以进一步确认构建模型的合理和可靠性,准确找出临界离解电场参数。
1 理论计算方法
本文采用不同的方法和基组,优化计算无外电场下MgH分子基态的几何结构和能量,计算结果与实验值进行比较,再结合能量最低原理[16],优选出B3P86方法和基组cc-PV5Z进行计算。沿分子z轴(H-Mg连线)方向,加一系列有限的外电场(-0.03~0.03 a.u.),优化计算和分析MgH分子的几何构型、偶极矩、振动频率和红外光谱强度与外加电场的变化关系。采用CCSD(T)方法,对不同外电场下MgH分子进行单点能扫描计算,利用软件作图获得势能曲线,全部计算均在Gaussian03程序包中完成。
采用Morse势函数拟合获得无外场下的势能函数,得出其势参数,在此基础上,利用偶极近似理论构建外电场下分子势能函数模型,编制程序拟合不同外电场下的势能曲线,得出拟合参数,与数值计算和理论分析进行比较,判断模型的合理和可靠性,准确得出临界离解电场参数。
2 解析势能函数模型
无外电场下,采用Morse势模型拟合势能曲线。其中,Morse势V是适用于稳定双原子分子的三参数函数[17]:
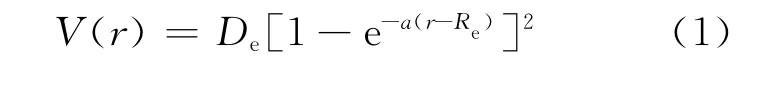
式中:De为离解能;a为Morse参量;r为核间距;Re为平衡核间距。
外电场作用下,分子体系能量的哈密顿量在无外电场的基础上,增加了外电场与分子体系的相互作用哈密顿量,H[18-19]变为:
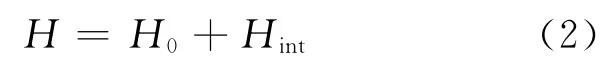
式中:H0为无外电场时的哈密顿量;Hint为外电场E与分子体系的相互作用哈密顿量。在偶极近似下,Hint可表示为:

式中,μ为分子电偶极矩。因此,外电场下分子的势能可分为无外电场的势能和外电场与分子间的相互作用势能,其标量表达式为:
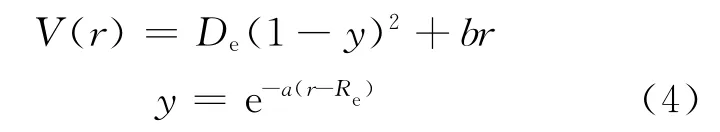
式中,b=-E(q+αE),为与外电场有关的量(相当于电场力),q为与固有偶极矩对应的偶极子电荷,α为外电场作用下产生的诱导偶极矩对应分子极化有关的电极化率参数。
3 结果和讨论
3.1 MgH分子的平衡几何和物理性质
MgH分子为线性双原子分子,属于C∞υ,沿分子z轴加上不同外电场,即正向电场(0~0.03 a.u.)和反向电场(-0.03~0 a.u.),采用B3P86和cc-PV5Z方法对MgH分子进行优化计算,得到分子稳定几何结构和物理性质参数。结果表明,MgH分子结构的对称性和电子组态不随外电场而改变,仍分别为C∞υ和2Σg,本文计算的无外电场下的偶极矩为1.389 3 D,与文献[20]的1.37 D较接近。键长、偶极矩、振动频率f和红外光谱(IR)强度随电场的变化如图1所示。从图1可看出,MgH分子的键长和偶极矩随正向电场增大先减小后增大,且幅度均不大,而随反向电场增大两者均大幅增大,特别当反向电场大于0.02 a.u.后,增大的幅度更大;振动频率随外电场的变化情况与前两者正好相反;红外光谱强度在正向电场中几乎不变,但在反向电场中变化的幅度较大。这些分析说明反向电场对分子固有电偶极矩的作用更大,极化现象更明显,导致分子的电荷分布、几何结构和物理性质参数变化幅度较大。
3.2 MgH分子势能函数
1)无外电场下MgH分子势能函数
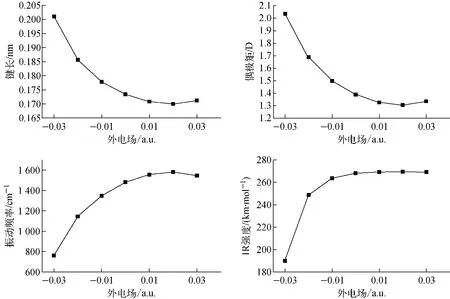
图1 键长、偶极矩、振动频率和红外光谱强度随外电场的变化Fig.1 Variation of bond length,dipole moment,vibration frequency and IR intensity with external electric field
无外电场时,采用CCSD(T)和cc-PV5Z方法对MgH分子进行单点能扫描,其中原子核间距变化步长为0.005 nm,共计算50个单点能,获得无外电场下的势能曲线。利用式(1)拟合无外电场下的势能曲线,得到势能函数解析表达式的各参数(表1),并与文献[10-12,19]和实验值[21]对比。从表1可看出,本文的拟合参数与实验值相差不大,相对误差分别为0.017%和0.26%,这些精确的参数用来拟合外电场作用下无外场势能函数部分是可靠的。
2)不同外电场下MgH分子势能函数
沿z轴分别加正向电场(0.01~0.04 a.u.)和反向电场(-0.01~-0.03 a.u.),采用相同方法进行单点能扫描,其中原子核间距变化步长为0.1 nm,共计算了30个单点能,不同外电场下的势能曲线示于图2,其中的小图为平衡位置附近势能图。从图中可看出,随着正向电场的增加,离解能缓慢下降,平衡核间距变化不大;而随着反向电场的增加,离解能逐渐减小,势能函数出现一稳定极小点和一非稳定极大点,类似“火山态”[17],极小和极大点之间的势垒减小,平衡键长增大。说明随着外电场的增加,离解能均减小,但反向电场减小较明显,即更易离解,这与物理性质参数变化分析一致,因此应从反向电场中找出临界离解电场参数。

表1 无电场下MgH分子势能函数拟合参数Table 1 Potential energy fitting parameters of MgH without external electric field
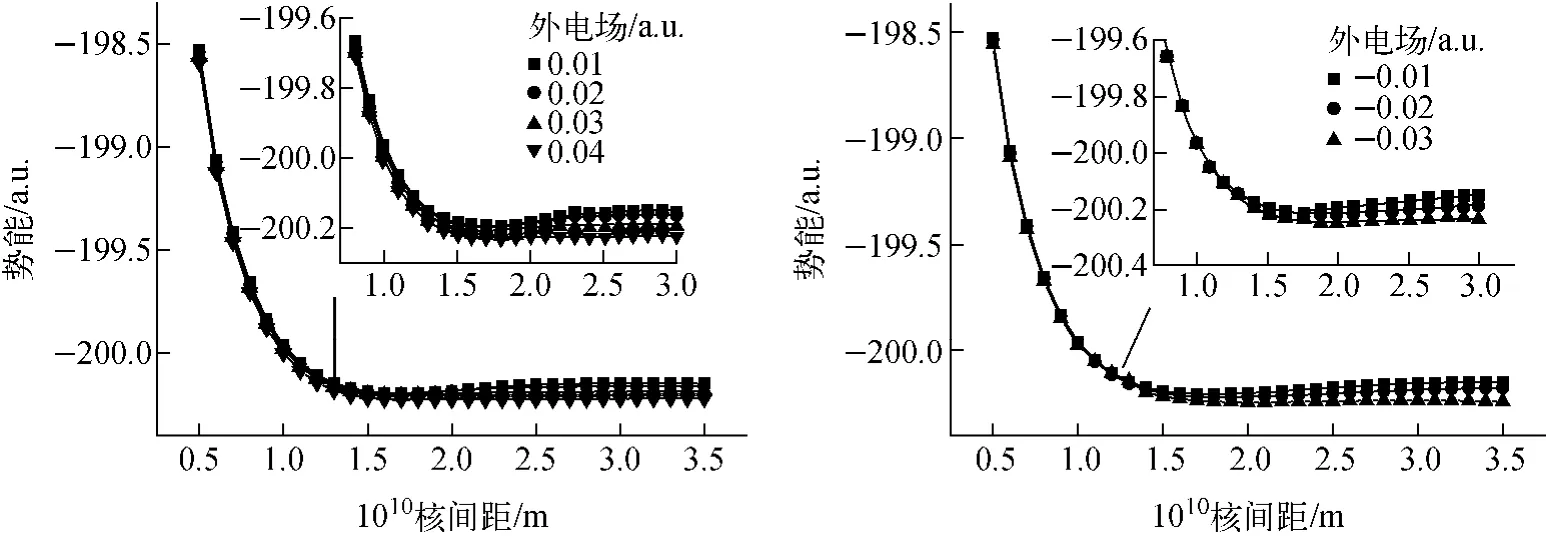
图2 不同外电场下MgH的势能曲线Fig.2 Potential energy curves of MgH in different external electric fields
为从数值计算中找出临界离解电场,本文进一步优化计算反向电场的分子结构,当反向电场设为-0.04、-0.038和-0.035 a.u.时,优化均不能进行,说明分子已发生离解,继续减小电场,当电场减为-0.034 a.u.时,优化才能进行。对外加-0.034 a.u.电场进行单点能扫描,势能曲线示于图3。从图可看出,当反向电场达-0.034 a.u.时,势能曲线的稳定点消失,势垒趋于0,分子开始发生离解,因此,MgH外电场下的临界离解电场为-0.034 a.u.,对应的优化离解键长为0.215 2 nm,偶极矩为2.297 6 D。
3)反向电场下MgH分子势能函数模型拟合参数
利用构建模型(式(4)),编制程序拟合临界电场的势能曲线,得出外电场参数b=-11.91 eV· nm-1,为计算临界电场参数,引入z=(b/2a)De,再由势能极值条件可得临界离解键长为:
Rc=Re+(1/a)ln 2=Re+0.693 1/a(5)
由于MgH为异核双原子分子,在外电场作用下分子的极化被忽略,则α=0,由离解条件可得到临界离解电场Ec为:

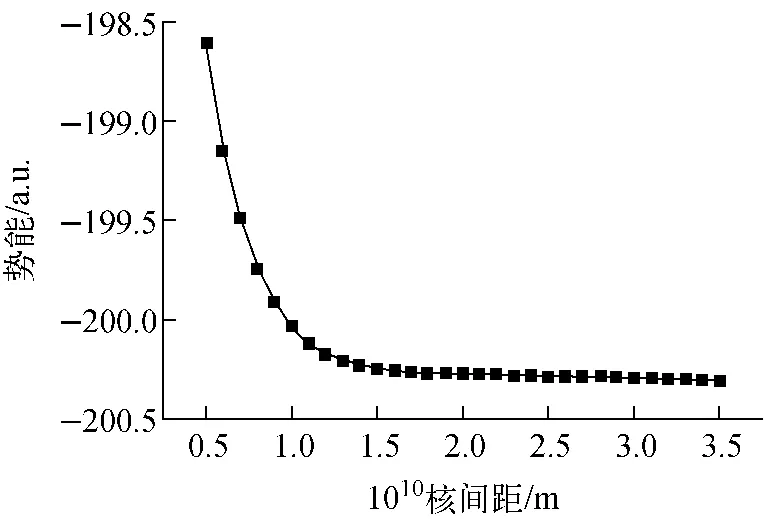
图3 临界离解电场下MgH的势能曲线Fig.3 Potential energy curve of MgH in critical dissociation electric field
由式(5)、(6)分别计算不同外电场下的临界离解键长和电场,结果表明,当外电场为-0.034 a.u.时,其临界离解电场和键长分别为-0.035 a.u.和0.213 4 nm,与数值计算结果较接近,相对误差仅2.94%和0.84%,这说明构建的模型是合理和可靠的。
4 结论
本文采用优选的密度泛函B3P86方法结合cc-PV5Z基组优化计算了不同外电场下MgH分子的几何结构和物理性质参数,同时采用能量高精度耦合簇CCSD(T)方法进行了单点能扫描计算。结果表明,平衡键长、偶极矩、振动频率和红外光谱强度随外电场的变化而变化,且在反向电场中变化幅度较大;随正向和反向电场的增加,势能曲线的离解能均下降,但外加反向电场时减小得更明显,平衡核间距变化均不大,出现一稳定极小点和一非稳定极大点,类似“火山态”,且极小和极大点之间的势垒减小,平衡键长增大,达到临界电场时,势能曲线的稳定点消失,极小和极大点之间的势垒趋于0,分子开始发生离解。利用Morse势模型拟合无外电场下的势能函数,得出势参数与实验值吻合较好,再采用构建的外电场中势模型编制程序拟合不同反向电场下势能函数曲线,得出相应的拟合参数,在此基础上计算出合理的临界离解电场参数,与数值计算相比,相对误差仅2.94%和0.84%,说明构建的外电场下势能函数模型是合理和可靠的。这为进一步研究外电场中分子光谱、动力学特性和分子Stark效应冷却提供重要的理论和实验参考。
[1]张中明,普小云,周曙白.MgO分子B1Σ+-X1Σ+带系及MgH分子A2Π-X2Σ+带系[J].原子与分子物理学报,1999,16(2):281-286.ZHANG Zhongming,PU Xiaoyun,ZHOU Shubai.Theoretical calculation of the oscillator strength of the B1Σ+-X1Σ+band system of MgO molecule and that of the A2Π-X2Σ+band system of MgH molecule[J].Journal of Atomic and Molecular Physics,1999,16(2):281-286(in Chinese).
[2]王秀丽,徐江平,张孝彬,等.掺Cr纳米晶Mg2Ni合金的气态储氢性能[J].中国有色金属学报,2002,12(5):907-911.WANG Xiuli,XU Jiangping,ZHANG Xiaobing,et al.Hydrogen storage properties of nanocrystal line Mg2Ni alloys with Cr additions[J].The Chinese Journal of Nonferrous Metal,2002,12(5):907-911(in Chinese).
[3]陈东,陈廉.Ca,Pd,Sn,La对MgH2电子结构的影响[J].稀有金属材料与工程,2004,33(5):485-489.CHEN Dong,CHEN Lian.Ca,Pd,Sn and La effects on the electronic structure of MgH2[J].Rare Metal Materials and Engineering,2004,33(5):485-489(in Chinese).
[4]BETHLEM H L,CROMPVOETS F M H,JONGMA R T,et al.Decreleration and trapping of ammonia using time-varying electric field[J].Physical Review A,2002,65(5):0534161.
[5]LEOPOLD K R,ZINK L R,EVENSON K M,et al.The far infrared spectrum magnesium hydride[J].Journal of Chemical Physics,1986,84:1 935-1 937.
[6]LEMOINE B,DEMUYNCK K,DESTOMBES J L,et al.Infrared diode laser spectra of MgH and MgD[J].Journal of Chemical Physics,1988,89:673-677.
[7]ZINK L R,JENNINGS A D,EVENSON K M,et al.Laboratory measurements for the astro-physical identification MgH[J].Astrophysics Journal of Letters,1990,359:65-69.
[8]MOSTAFANEJAD M,SHAYESTEH A.ab initio potential energy curves and transition dipole moments for the X2Σ+,A2Πand B2Σ+states of MgH[J].Chemical Physics Letters,2012,551:13-18.
[9]MESTDAGH J M,PUJO P,SOEP B,et al.ab initio calculation of the ground and excited states of MgH using pseudopotential approach[J].Chemical Physics Letters,2009,471:22-28.
[10]宇燕.MgH和MgD分子的结构与势能函数[J].四川大学学报:自然科学版,2006,43(6):1 332-1 336.YU Yan.Potential energy function of MgH,MgD molecule[J].Journal of Sichuan University:Natural Science,2006,43(6):1 332-1 336(in Chinese).
[11]吕兵,令狐荣锋,杨向东.MgH分子X2Σ+,A2Π电子态的势能函数[J].四川大学学报:自然科学版,2008,45(1):125-130.LV Bing,LINGHU Rongfeng,YANG Xiangdong.Analytical potential function for the electronic states X2Σ+,A2Πof MgH molecule[J].Journal of Sichuan University:Natural Science,2008,45(1):125-130(in Chinese).
[12]张萍,张忠.MgHn+(n=0,1)分子及分子离子的结构与性质研究[J].四川师范大学学报:自然科学版,2005,28(5):601-603.ZHANG Ping,ZHANG Zhong.Study on the structure and property of MgHn+(n=0,1)[J].Journal of Sichuan Normal University:Natural Science,2005,28(5):601-603(in Chinese).
[13]黄多辉,王藩侯,万明杰,等.外场下SnS分子结构及其特性[J].物理学报,2013,62(1):013104.HUANG Duohui,WANG Fanhou,WAN Mingjie,et al.SnS molecular structure and properties under external electric field[J].Acta Physica Sinica,2013,62(1):013104(in Chinese).
[14]伍冬兰,涂娟,万慧军,等.CH自由基在外电场中的分子结构和势能函数[J].原子与分子物理学报,2014,31(2):197-201.WU Donglan,TU Juan,WAN Huijun,et al.Molecular structure and potential energy function of CH molecule under external electric field[J].Journal of Atomic and Molecular Physics,2014,31(2):197-201(in Chinese).
[15]伍冬兰,涂娟,万慧军,等.外电场下BH分子势能函数[J].计算物理,2014,31(1):115-120.WU Donglan,TU Juan,WAN Huijun,et al.Potential energy functions of BH molecule in external electric fields[J].Journal of Compute Physics,2014,31(1):115-120(in Chinese).
[16]朱正和.原子分子反应静力学[M].北京:科学出版社,1996.
[17]朱正和,俞华根.分子结构与分子势能函数[M].北京:科学出版社,1997.
[18]徐国亮,朱正和,马美仲,等.甲烷激发态外场效应[J].物理学报,2005,54(7):3 087-3 093.XU Guoliang,ZHU Zhenghe,MA Meizhong,et al.Study on the effect of external electric field excitation on methane[J].Acta Physica Sinica,2005,54(7):3 087-3 093(in Chinese).
[19]CHAUDHURI R K,MUDHOLKAR A,FREED K F,et al.Application of the effective valence shell Hamiltonian method to accurate estimation of valence and Rydberg states oscillator strengths and excitation energies forπelectron systems[J].Journal of Chemical Physics,1997,106:9 252-9 264.
[20]BRUNA P J,GREIN F.Hyperfine coupling constants,electron-spin g-factors and vertical spectra of the radicals Be H,MgH,Ca H and BZ+,AlZ+,GaZ+(Z=H,Li,Na,K):A theoretical study[J].Physical Chemist Chemical Physics,2003,5:3 140-3 153.
[21]HUBER K P,HERZBERG G.Molecular spectra and molecular structureⅣ:Constants of diatomic molecules[M].New York:Van Nostrand Reinhold Company,1978.
Change Law of MgH Molecular Characteristics and Potential Energy with External Electric Field
WU Dong-lan,TAN Bin,WEN Yu-feng,WAN Hui-jun,XIE An-dong
(College of Mathematics and Physics,Jinggangshan University,Ji’an 343009,China)
Adopting density functional method B3P86 and cc-PV5Z and setting different electric fields,the geometric structures of MgH molecule were optimized,and the bond lengths,dipole moments,vibration frequencies,IR intensities and other physical property parameters were obtained.Using the energy of high precision coupled cluster method CCSD(T)and the same basis set to scan single point energies,the potential energy curves of different external fields were gotten.The results show that the physical property parameters and potential energy values change with the external electric fields,especially at reverse direction electric fields.In order to get the critical dissociation electric parameters,the dipole approximation was adopted to construct potential energy function model,then the model was put to fit the corresponding potential energy curves of external electric fields.It is found that the fitted critical dissociation electric parame-ters are consistent with the numerical calculation values and theoretical analysis results,and the relative errors are less than 3%,so the constructed model is reliable and accurate.These will provide important theoretical and experimental reference for further studying the molecular spectroscopy,molecular dynamics and molecular cooling with Stark effect.
MgH molecule;potential energy function model;external electric field
O561.3
:A
:1000-6931(2015)12-2118-06
10.7538/yzk.2015.49.12.2118
2014-10-09;
:2015-03-16
国家自然科学基金资助项目(11147158,11264020);江西省自然科学基金资助项目(2010GQW0031);江西省教育厅科技项目资助(GJJ12483,GJJ12463,GJJ11540)
伍冬兰(1978—),女,江西吉安人,副教授,博士,从事分子势能函数与光谱分析研究
