聚天冬氨酸对CaCO3的阻垢作用及其分子动力学模拟
2015-07-02冷一欣
韶 晖,周 胤,王 雅,冷一欣,钟 璟
(常州大学 石油化工学院 江苏省绿色催化材料与技术重点实验室,江苏 常州 213164)
聚天冬氨酸对CaCO3的阻垢作用及其分子动力学模拟
韶 晖,周 胤,王 雅,冷一欣,钟 璟
(常州大学 石油化工学院 江苏省绿色催化材料与技术重点实验室,江苏 常州 213164)
以氨水与马来酸酐为原料,热缩聚合成了聚天冬氨酸,表征结果显示其结构中含有α和β两种构型。通过静态阻垢实验测定了PASP对碳酸钙的阻垢性能,当PASP投加量为10 mg/L时,阻垢率达到96.9%。采用分子动力学方法模拟计算了分子态和离子态PASP与方解石(104)和(110)晶面的相互作用,结果表明,离子态PASP-(110)晶面结合能、分子态PASP-(110)晶面结合能、离子态PASP-(104)晶面结合能、分子态PASP-(104)晶面结合能依次降低,说明PASP对方解石(110)晶面生长的抑制居于主导地位,与实验得到的结论相一致。
聚天冬氨酸;合成;阻垢性能;方解石;分子动力学
随着水资源的短缺和人类环保意识的增强,可生物降解的绿色环保型阻垢剂已成为当今水处理剂的研究方向。聚天冬氨酸(PASP)是一种带有羧酸侧链的聚氨基酸,具有优异的阻垢分散性和较强的生物降解性,是公认的绿色聚合物和水处理剂的更新换代产品。虽然,对聚天冬氨酸的研究取得了很大的进展,但工作仅限于合成、复配、性能评定和应用方面。
分子动力学从分子结构的角度对阻垢剂的作用机理进行研究,解释阻垢剂与成垢晶体相互作用的本质。目前,采用分子动力学(MD)方法研究阻垢机理较多的为聚合物阻垢剂。夏明珠等[1]采用MD方法模拟了有机膦酸聚合物与方解石晶面的相互作用。Shi等[2]研究了聚环氧琥珀酸(PESA)、丙烯酸共聚物等水溶性聚羧酸与方解石晶体主要生长面的相互作用,并探讨了相互作用的本质。Zhang等[3]对CaCO3晶体和低聚物马来酸、丙烯酸等之间的相互作用进行了分子模拟。采用MD方法对PASP进行模拟研究仅有少量报道。Zeng等[4]采用MD方法,在真空条件下构建PASP与CaCO3垢晶体主要生长面的分层模型,根据计算的结合能、形变能等分析了其阻垢性能的强弱。在本研究中,将实验与模拟相结合,首先以氨水与马来酸酐为原料,热缩聚合成PASP,采用静态阻垢法测定其对CaCO3的阻垢性能;然后采用MD方法模拟计算PASP与方解石(110)和(104)晶面的相互作用。
1 实验部分
1.1 主要试剂
氢氧化钠、马来酸酐、氨水、碳酸氢铵、盐酸,AR,国药集团化学试剂有限公司产品;尿素,AR,上海润捷化学试剂有限公司产品;碳酸铵,江苏强盛功能化学股份有限公司产品。
1.2 聚天冬氨酸的制备
向三口烧瓶中加入马来酸酐(MA)和去离子水,加热搅拌0.5 h;冷却至室温后,以n(MA)/n(NH3(H2O)=1/1.4的比例滴加氨水,80℃下氨化2 h;减压蒸馏后,在220℃下聚合8 h,得棕红色物体;滴加2 mol/L氢氧化钠溶液,调节pH值为9.0,室温水解1.0 h;用0.1 mol/L的稀盐酸调至中性,加入适量的无水乙醇,搅拌,产生红棕色沉淀,经过滤、真空干燥,得到聚天冬氨酸(PASP)。
1.3 性能表征
采用美国Nicolet公司PROTÉGÉ 460型傅里叶变换红外光谱仪测定样品的红外光谱(FT-IR),KBr压片;采用瑞士Bruker公司AVANCE III 400M核磁共振仪测定样品的核磁共振氢谱(1H-NMR);采用日本 Rigaku公司D/max 2500 PC型X射线衍射(XRD)仪测定CaCO3的物相。
2 结果与讨论
2.1 制备的聚天冬氨酸(PASP)的分子结构
图1、图2分别为所制备的PASP的FT-IR和1H NMR谱。图1显示,所制备的PASP的FT-IR谱中的吸收峰与文献值[5]一致,说明所合成的产物为聚天冬氨酸。

图1 所制备的PASP的FT-IR谱
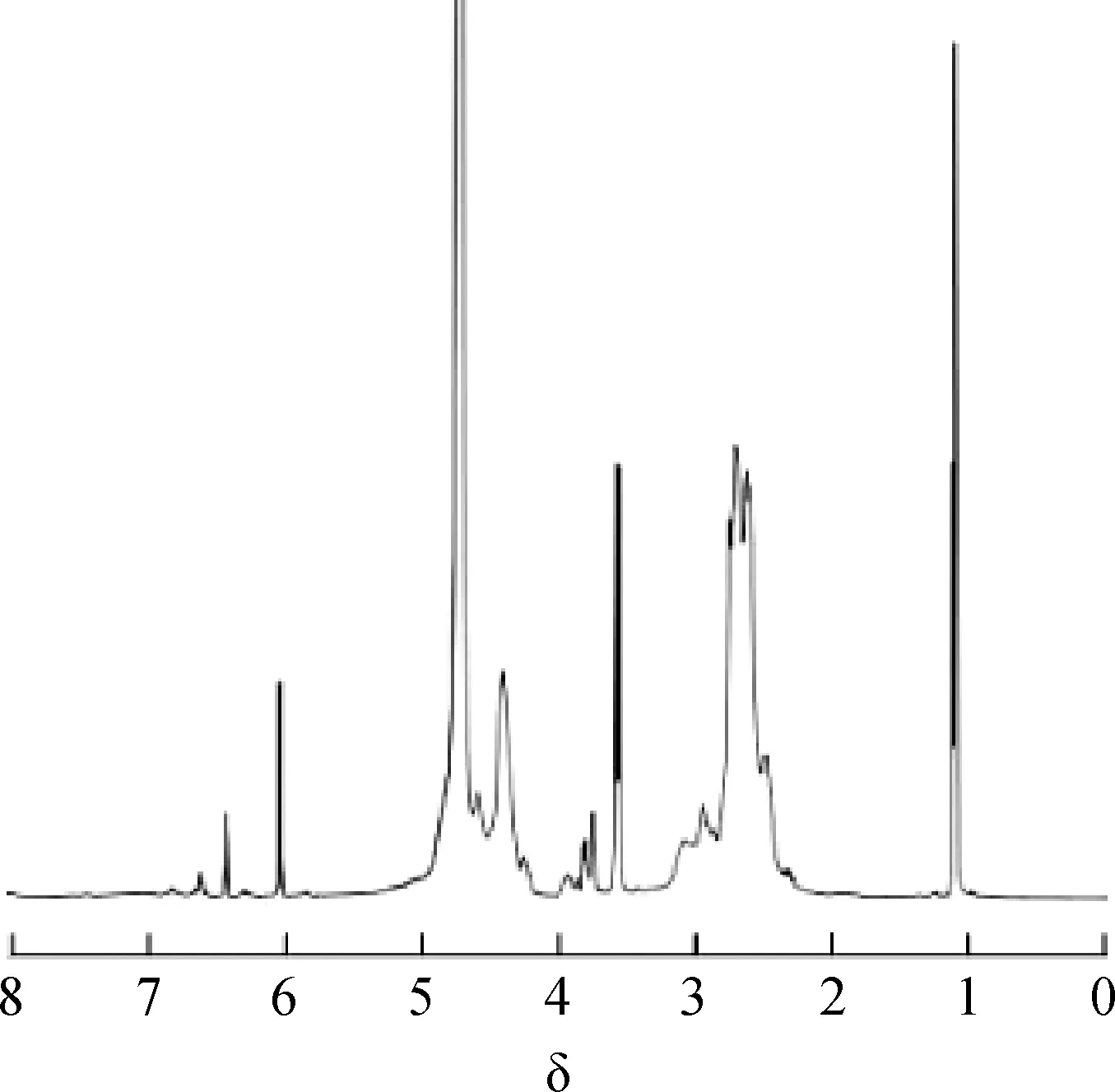
图2 所制备的PASP的1H-NMR谱
图2所示1H NMR谱中,δ=4.700处为溶剂重水(D2O)峰,δ在2.576~2.717范围的为亚甲基[—CH2]上H核产生的共振峰,δ在4.384~4.569范围的为次甲基[—CH—]中的H核产生的共振峰,表明共聚物结构中有α和β两种构型,其分子结构如图3 所示[6],δ=4.384为β构型中—CH—的H质子峰,δ=4.569为α构型中—CH—的H质子峰。由积分面积计算出β构型约占75%,α构型约占25%。

图3 由1H NMR得出的所制备的PASP的分子结构式
2.2 所制备的PASP的阻垢性能

图4 PASP添加量对CaCO3阻垢率的影响
实验中,对CaCO3垢体进行XRD分析,探讨PASP对CaCO3晶型的影响,结果如图5所示。
由图5可知,未加PASP所形成的CaCO3垢体以方解石晶型稳定存在,2θ在29.4°处(方解石(104)晶面)有很强的衍射峰,35.9°处(方解石(110)晶面)的衍射峰次之,同时在39.4°、43.1°、47.5°和48.5°处有衍射峰(分别归属于方解石(113)、(202)、(108)和(116)晶面),说明生成的垢体为方解石晶体,且(104)晶面和(110)晶面是它的主要生长面[7]。加入阻垢剂PASP后,CaCO3垢体的晶型发生了变化,2θ在26.2°、27.2°和31.4°处出现新的衍射峰,分别对应球霰石晶型的(100)、(101)和(102)晶面,即CaCO3以方解石和球霰石两种晶型的混合物存在。2θ在29.4°和35.9°处的衍射峰强度明显减弱,说明PASP能抑制方解石晶体(104)和(110)晶面的生长,且更能有效地抑制(110)晶面的生长。

图5 CaCO3垢体的XRD谱
3 PASP对CaCO3的阻垢作用的MD模拟
3.1 模拟方法
采用Material Studio4.4程序中的Discover模块,使用COMPASS力场对PASP的阻垢作用进行MD模拟。
采用切割分面的模式,研究PASP与热力学最稳定的CaCO3晶型(方解石晶体的相互作用机理。运用Material Studio4.4程序中的visualizer模块构建方解石晶胞(晶胞参数为a=b=0.4988 nm,c=1.7061 nm,α=β=90°,γ=120°[8])。

冷却水的pH值为8.0~9.0,为弱碱性, PASP中的部分羧基会电离成羧酸根,故在本研究中考察了分子态PASP和离子态PASP与方解石晶体的相互作用。取PASP的聚合度为20,XRD表征结果显示α和β两种构型含量比为1∶3,以嵌段共聚的方式构建分子态PASP及其中有1个羧基发生电离的离子态PASP,如图6所示。

图6 分子态及电离态PASP的结构式
PASP中的单体会发生扭曲、旋转而导致构型不同,能量也不同。为了使模拟结果接近实际,取单体扭转角度分别为0°、±45°、±90°、±135°和180°,构建PASP的8种构型,对其进行初步模拟。模拟条件为温度350 K、NVT系综、模拟时间100 ps、步长1 fs,每5000步输出1次结果,总共20帧。保存最后1帧聚合物的构型,并采用分子力学方法(MM)对其进行能量最优化[3],选择能量最低的构型作为MD模拟的初始构型。经比较MM能量最优化的聚合物的能量发现,扭转角度为135°时构建的聚合物的能量较低,构型稳定,如图7所示。
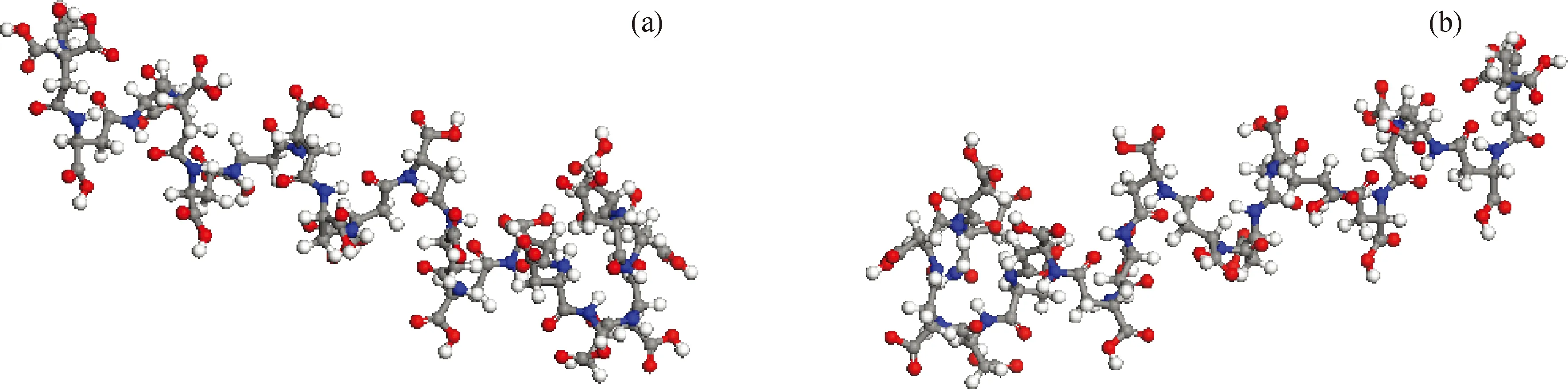
图7 分子态和离子态PASP的最优构型
分别将分子态及离子态PASP的最优构型放置于方解石晶体的(110)和(104)晶面,真空层厚度2.5 nm,采用NVT系综和Berendsen恒温器,根据Maxwell-Boltzmann分布确定各分子或离子的初始速度,运用Velocityverict法积分求和。根据0.95 nm的球形截断值计算体系非键作用,同时采用平均密度近似方法校正截断距离之外的分子间相互作用能[11]。模拟退火中,取1050 K为初始温度,350 K为最终温度,每50 K降温1次。依次在各温度点上进行MD模拟,共15次,平衡后取样分析。模拟退火由自编程序进行,模拟步长为1 fs,模拟时间为200 ps(体系平衡和取样分析各100 ps),每100步记录1次体系的轨迹[2]。
3.2 模拟结果与讨论
3.2.1 PASP与方解石晶体相互作用的平衡依据
在MD模拟过程中,根据温度和能量来判断体系是否达到平衡[12]。图8和图9分别为分子态和离子态PASP与方解石晶体相互作用模拟体系的温度变化。可见各个体系的温度波动范围不大,趋于平缓,说明体系温度已达平衡。
图10和11给出了分子态和离子态PASP与方解石晶体相互作用的能量波动。结果显示,各个体系的能量波动平缓,表明模拟体系的能量已达到平衡状态。
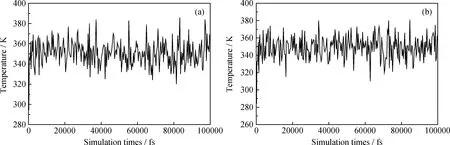
图8 分子态PASP与方解石晶体(110)和(104)面结合的温度波动
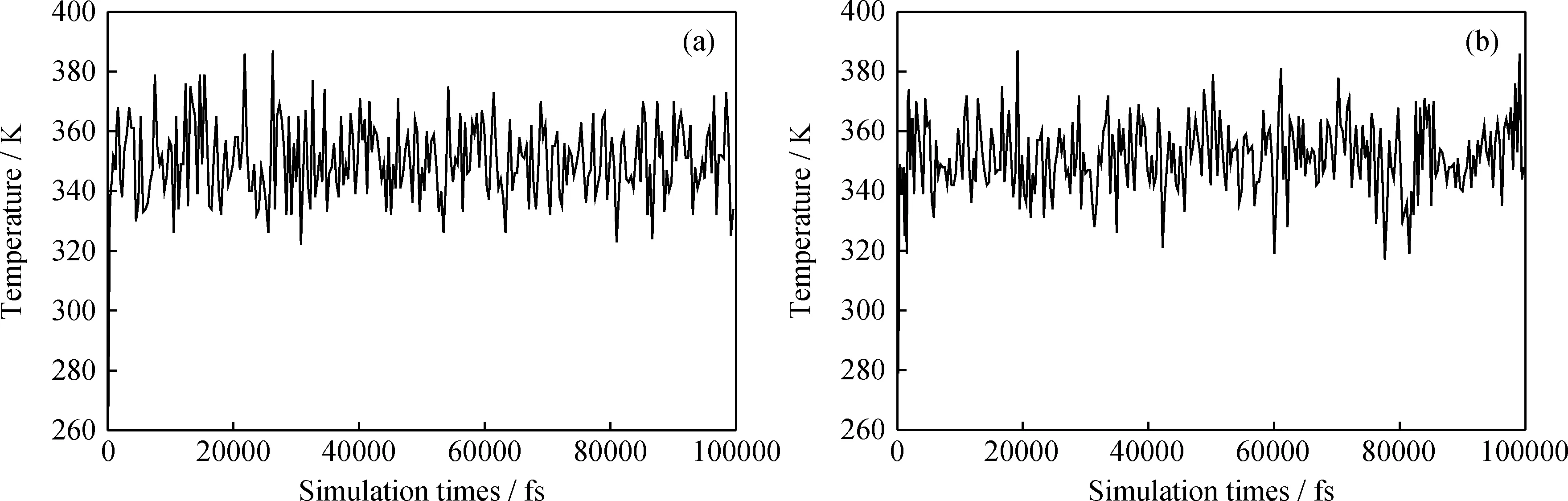
图9 离子态PASP与方解石晶体(110)和(104)面结合的温度波动
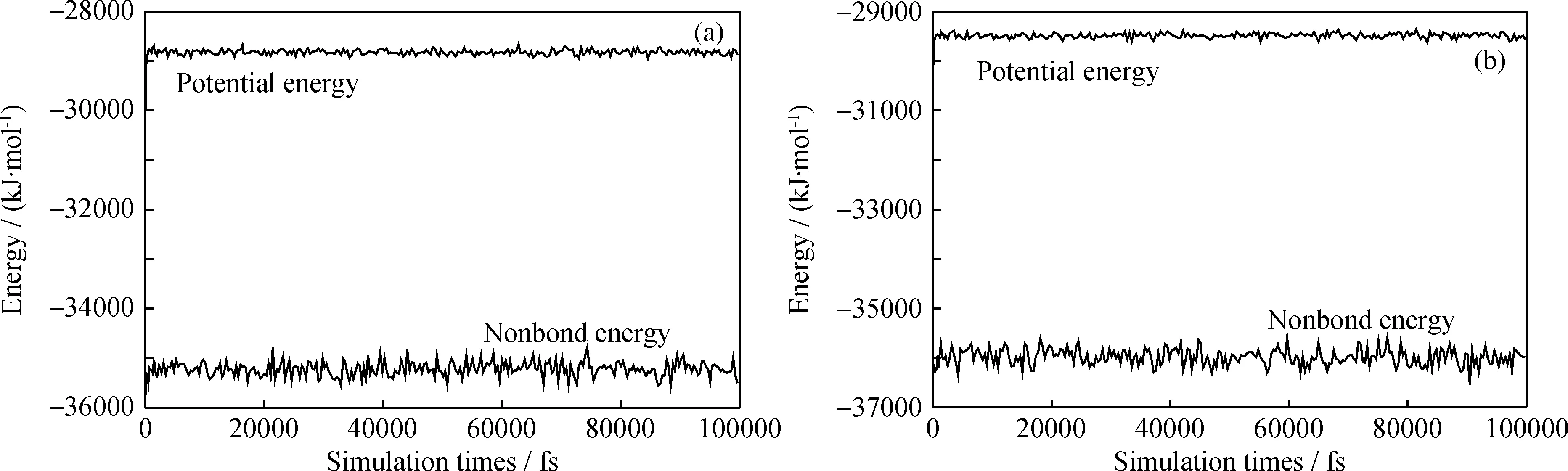
图10 分子态PASP与方解石晶体(110)及(104)面相互作用的能量波动

图11 离子态PASP与方解石晶体(110)及(104)面相互作用的能量波动
通过以上温度和能量判据,表明分子态PASP和离子态PASP与方解石晶体构成的超分子体系均已达到平衡,由此可见模型得到的分析数据可靠。
3.2.2 PASP在方解石晶面上的结合能
ΔE=Ecomplex-Epolymer+surface
(1)
式(1)中,Ecomplex为MD模拟200 ps后超分子体系的能量;Epolymer+surface为MD模拟前超分子体系的能量。
超分子体系达到平衡状态后,以步长1 fs、模拟时间100 ps、每100步记录1次体系轨迹进行模拟,收集体系的全轨迹,结果示于图12和图13,能量计算结果列于表1和表2。
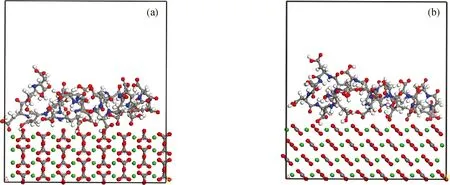
图12 MD模拟分子态PASP与方解石晶体(110)及(104)面的相互作用

图13 MD模拟离子态PASP与方解石晶体(110)及(104)面的相互作用
表1 PASP与方解石两晶面的结合能和形变能
Table 1 Binding and deformation energies between PASP and two crystal surfaces of calcite kJ/mol

CleavedsurfacePolymerEcomplexEpolymer+surfaceΔEEbindEpolymerEpolymer⁃bindΔEdeform110MolecularPASP-31032 73-29751 68-1281 051281 05-5783 79-5636 98146 81IonicPASP-31791 53-30488 05-1303 481303 485992 416120 82128 41104MolecularPASP-29644 35-28492 50-1151 851151 85-5783 79-5608 15175 64IonicPASP-30326 43-29162 81-1163 621163 625992 416130 73138 32

表2 PASP与方解石两晶面的非键相互作用能
表2列出的PASP与方解石两晶面的非键相互作用能为库仑相互作用能和范德华作用能之和。从表2可以看出,分子态及离子态PASP与方解石晶体晶面的非键相互作用能均为负值,其绝对值大小的顺序与表1所列结合能的排列顺序相同。由于PASP与方解石吸附为放热过程,非键相互作用能为负值,说明促进了PASP与方解石的结合[14];非键相互作用能越负,即其绝对值越大,PASP与方解石晶面的静电作用就越大,二者的结合程度也随之增大。由此得出,离子态PASP更易与方解石(110)晶面发生吸附作用,与以上结合能分析结果一致。
从表2还可以看出,ΔEcoulomb的绝对值远远大于ΔEvdw,表明PASP与方解石晶体形成超分子体系的过程中,库仑相互作用能起到决定性的作用,这与石文艳等[15]所得结论一致。
3.2.3 PASP在方解石晶面上的形变能
在聚合物阻垢剂和方解石相互作用的过程中,阻垢剂会发生一定程度的形变,其形变程度由形变能的大小反映。由式(2) 可计算得到形变能。
ΔEdeform=Epolymer-bind-Epolymer
(2)
式(2)中,Epolymer-bind和Epolymer分别为PASP聚合物在束缚和自由状态下的单点能[15]。
由式(2)计算得到的分子态和离子态PASP与方解石(110)和(104)晶面的形变能已列于表2。表2数据显示,无论是分子态PASP还是离子态PASP,均与方解石的两晶面发生了一定的作用,导致聚合物产生扭曲变形。从同一种PASP聚合物与方解石某一晶面的相互作用能可以看出,二者的非键相互作用能的绝对值远远大于聚合物的形变能,表明PASP聚合物具有克服自身扭曲变形的能力,同时还可以与方解石发生吸附作用,从而影响方解石晶体的正常生长,达到阻垢目的。
4 结 论
(1)以氨水与马来酸酐为原料,热缩聚合可得到阻垢剂聚天冬氨酸(PASP),其分子结构包括α和β两种构型;当PASP投加量为10 mg/L时,对CaCO3的阻垢率达到96.9%。
(2)PASP与方解石的分子动力学模拟结果表明,PASP对方解石(110)面生长的抑制作用居于主导地位;库仑相互作用能ΔEcoulomb均为负值,促进聚合物与方解石的结合,且ΔEcoulomb的绝对值远远大于范德华作用能ΔEvdw,在PASP与方解石形成超分子体系的过程中起决定性的作用;PASP与方解石相互作用过程中,在克服自身扭曲变形的同时,还可以与方解石发生吸附作用,从而影响方解石晶体的正常生长。
[1] 夏明珠, 雷武, 戴林宏, 等. 膦系阻垢剂对碳酸钙阻垢机理的研究[J].化学学报, 2010, 68(2): 143-148.(XIA Mingzhu, LEI Wu, DAI Linhong, et al.Study of the mechanism of phosphonate scale inhibitors against calcium carbonate scale[J].Acta Chimica Sinica, 2010, 68(2): 143-148.)
[2] SHI W Y, DING C, YAH J L, et al.Molecular dynamics simulation for interaction of PESA and acrylic copolymers with calcite crystal surfaces[J].Desalination, 2012, 291(2):8-14.
[3] ZHANG Q, REN H, WANG W, et al.Molecular simulation of oligomer inhibitors for calcite scale[J].Particuology, 2012, 10(3): 266-275.
[4] ZENG J P, WANG F H, GONG X D.Molecular dynamics simulation on the interaction between polyaspartic acid and calcium carbonate[J].Molecular Simulation, 2013, 39(3): 169-175.
[5] 中华人民共和国国家发展和改革委员会. HG/T3822-2823-2006.聚天冬氨酸(盐)[S].北京: 化学工业出版社.2007-03-01.
[6] 李安生. 聚天冬氨酸的合成及其与金属离子络合沉淀的研究[D].西安:西北大学, 2005.
[7] 赵长林. IA-AMPS-HPA三元共聚物的合成及阻垢性能、机理的研究[D].南京:南京理工大学, 2010.
[8] YUAN P Q, KONG N, CHENG Z M, et al.Electrostatic potential on anti-scalants modified CaCO3(104) surface: A molecular simulation study[J].Desalination, 2009, 238(1): 246-256.
[9] RAI B, RAO T K, KRISHNAMURTHY S, et al.Molecular modeling of interactions of alkyl hydroxamates with calcium minerals[J].Journal of Colloid and Interface Science, 2002, 256(1): 106-113.
[10] PRADIP, RAI B, RAO T K, et al.Molecular modeling of interactions of diphosphonic acid based surfactants with calcium minerals[J].Langmuir, 2002, 18(3): 932-940.
[11] 石文艳, 王风云, 夏明珠, 等.羧酸共聚物与方解石晶体相互作用的 MD 模拟[J].化学学报, 2006, 64(17): 1817-1823.(SHI Wenyan, WANG Fengyun, XIA Mingzhu, et al.Molecular dynamics simulation of interaction between carboxylate copolymer and calcite crystal[J].Acta Chimica Sinica, 2006, 64(17): 1817-1823.)
[12] Frenkel & Smit,著, 汪文川, 等译.分子模拟—从头算法到应用[M].北京:化学工业出版社.2002.
[13] 王世燕, 张军, 卢贵武.聚合物阻垢机理的分子动力学模拟研究[J].中国石油大学学报: 自然科学版, 2007, 31(5): 144-147.(WANG Shiyan, ZHANG Jun, LU Guiwu.Molecular dynamics simulation on mechanism of scale inhibition of polymer[J].Journal of China University of Petroleum, 2007, 31(5): 144-147.)
[14] 曾建平. 无磷阻垢缓蚀剂的分子动力学模拟研究[D].南京:南京理工大学, 2013.
[15] 石文艳, 吕志敏, 王风云, 等.聚环氧琥珀酸及丙烯酸共聚物阻垢机理的分子动力学模拟[J].化学通报, 2011, 74(7): 651-658.(SHI Wenyan, LÜ Zhimin, WANG Fengyun, et al.Molecular dynamics simulation of scale inhibiting mechanism of PESA and acrylic copolymer[J].Chemistry Online, 2011, 74(7): 651-658.)
Scale Inhibiting Effect and Molecular Dynamics Simulation of Polyaspartic Acid on CaCO3
SHAO Hui, ZHOU Yin, WANG Ya, LENG Yixin, ZHONG Jing
(JiangsuKeyLaboratoryofAdvancedCatalyticMaterialsandTechnology,SchoolofPetrochemicalEngineering,ChangzhouUniversity,Changzhou213164,China)
With ammonia and maleic anhydride as raw materials, polyaspartic acid (PASP) was synthesized by thermal polycondensation. The characterization results showed that PASP containedαandβtwo kinds of configuration. The scale inhibition of PASP on CaCO3was determined through the static anti-scaling method. When the dosage of PASP was 10 mg/L, the scale inhibition rate of CaCO3was 96.9%. At the same time, the interactions of molecular and ionic PASP with (110), (104) crystal surfaces of calcite were simulated by molecular dynamics (MD). The results showed that the binding energy for polymers with two calcite crystal surface were decreased in the order of those for ionic PASP, molecular PASP with (110) crystal surface,and then ionic PASP,molecular PASP with (104) crystal surface, indicating that the combination of PASP with (110) crystal surface was more firmly than those with (104) crystal surface, so the scale-inhibition of PASP on the growth of (110) crystal surface was dominant, which was consistent with experimental conclusion.
polyaspartic acid(PASP); synthesis; scale inhibition; calcite; molecular dynamics (MD)
2014-06-00
江苏省产学研前瞻性联合研究项目(BY2014037-15)资助
韶晖,女,副教授,博士,主要从事水处理剂的制备和分离技术;E-mail:shaohui200800@cczu.edu.cn
1001-8719(2015)05-1203-08
TQ085
A
10.3969/j.issn.1001-8719.2015.05.025
