综述原位聚合法制备高性能聚乙烯基复合材料
2015-06-24何富安张黎明
何富安, 张黎明
(1.中山大学 化学与化学工程学院 高分子研究所 PCFM和GD HPPC实验室, 广东 广州 510275;2.广东石油化工学院 化学工程学院高分子材料系, 广东 茂名 525000)
综述原位聚合法制备高性能聚乙烯基复合材料
何富安1,2, 张黎明1
(1.中山大学 化学与化学工程学院 高分子研究所 PCFM和GD HPPC实验室, 广东 广州 510275;2.广东石油化工学院 化学工程学院高分子材料系, 广东 茂名 525000)
综述了近年来国内外通过原位聚合法制备高性能聚乙烯基复合材料的研究进展,主要涉及黏土/聚乙烯复合材料、碳纳米管/聚乙烯复合材料、石墨类填料/聚乙烯复合材料等体系,重点介绍了相关材料制备时所用负载型催化剂的聚合反应规律,以及所得聚乙烯基复合材料的形貌、结构与性能,还指出了该领域未来研究面临的一些挑战。
聚乙烯;复合材料;原位聚合;改性剂
作为五大合成树脂之一,聚乙烯不仅具有良好的化学稳定性、韧性和电绝缘性,而且具有耐侯、耐低温、不易吸水等优点。然而,单一聚乙烯材料的尺寸稳定性、耐热性、耐环境应力开裂性及阻隔性较差,且其韧性有余而刚性和强度不足。因此,采用增强剂并探索有效途径与方法对聚乙烯进行复合改性,具有重要意义。此外,将聚乙烯与具有光、电、磁等特殊功能的填料复合,还可实现聚乙烯材料的功能化,从而扩展其使用范围。
迄今为止,制备聚乙烯基复合材料的方法主要有溶液共混、熔融共混和原位聚合[1]。其中,原位聚合方法一般是将配位催化剂首先负载到填料上面,然后以负载催化剂催化乙烯进行聚合反应,最终就地生成聚乙烯基复合材料。与其它2种方法相比,原位聚合法具有以下4个优点[2]。(1)只需经过一步聚合反应就可以在得到聚乙烯的同时得到复合材料;(2)填料分散比较均匀,且与聚乙烯基体之间的界面相互作用较强;(3)能够制备高填充和超高相对分子质量的聚乙烯基复合材料,这是通过熔融共混和溶液共混都较难实现的,而且高填充聚乙烯基复合材料还可作为母料,进一步与其它聚合物熔融共混制备相应的复合材料;(4)由于使用的是负载催化剂,适用于现有的聚乙烯工业生产设备。因此,原位聚合一直是制备聚乙烯基复合材料的热门方法。原位聚合制备聚乙烯基复合材料所使用的催化剂主要是Zeigler-Natta催化剂、茂金属催化剂和后过渡金属催化剂,所用的填料主要有黏土、碳纳米管、石墨类填料、水滑石、二氧化硅、磁性粒子、二氧化锆等[3-92]。笔者结合自己的相关研究工作,以不同填料改性剂为线索,综述了近年来国内外通过原位聚合制备高性能聚乙烯基复合材料的最新研究进展。
1 黏土/聚乙烯复合材料
在通过原位聚合制备聚乙烯基复合材料的研究中,黏土/聚乙烯复合材料是研究最广泛的体系。无机黏土作为层状硅酸盐(其典型的2∶1结构如图1所示),包括了蒙脱土、蛭石、海泡石、凹凸棒等多种类型[1]。无机黏土的片层厚度大约在1 nm左右,由硅、镁、铝、氧、氢等元素成,层与层之间有可交换的水合阳离子。黏土自身的结构特点决定了要成功地通过原位聚合法制备黏土/聚乙烯基复合材料,需要考虑催化剂是否能够负载到黏土片层之间,以及对无机黏土进行有机插层改性两个关键因素。如果催化剂只是负载于黏土的表面,在聚合反应开始后,聚乙烯分子链仅能在黏土表面进行包裹,并不能使黏土片层有效剥离,因而不能提高甚至可能会降低材料的性能;如果催化剂能有效地负载于黏土片层之间,那么在聚合过程中,乙烯单体小分子进入到黏土片层之间与催化剂活性中心进行反应,随着反应的不断进行,生成的聚乙烯分子链不断增长,当其尺寸增大到超过黏土片层层间距离时,聚合作用力大于黏土片层之间的库仑引力,从而将黏土片层逐渐推开,最终使其剥离开来并均匀分布于聚乙烯基体之中。另外,无机黏土与有机非极性聚乙烯基体的相容性差,用有机改性剂对无机黏土进行插层改性,既能提高黏土与聚乙烯基体的界面相互作用,又能增大黏土的层间距,使得在聚合过程中乙烯单体小分子更容易进入黏土片层间进行反应。
制备黏土/聚乙烯复合材料的最终目的是为了提高聚乙烯材料的性能或实现其功能化,因此所得复合材料的微观结构及其对材料综合性能的影响是科研工作者最关注的问题。与此同时,由于原位聚合法需要将催化剂负载在黏土片层之间催化聚合反应,因此黏土不仅仅是作为纳米增强剂的原材料,同时在聚合过程中还为催化剂提供了1个纳米级尺寸的聚合反应受限空间。这实质上是个负载催化剂催化乙烯进行聚合的过程,其中必然涉及到负载催化剂的催化性能、各种聚合条件对聚合反应的影响、聚合反应动力学等问题。这些问题同样是科研工作者所研究的重点。
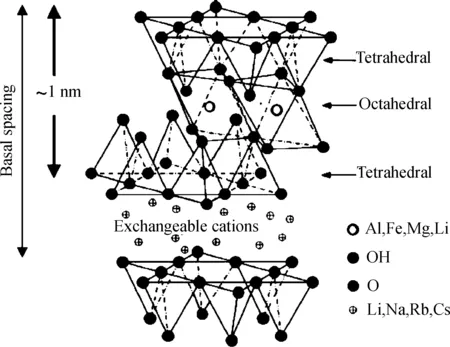
图1 2∶1层状硅酸盐结构[1]
1.1 黏土负载Zeigler-Natta催化剂体系
黏土负载Zeigler-Natta催化剂分为单组分载体和复合载体2个体系。复合载体主要由黏土和含镁化合物组成,而其中的含镁化合物则是工业Zeigler-Natta催化剂的重要载体组分。使用复合载体的目的是为了克服单组分黏土载体对乙烯聚合活性中心的毒害作用,从而维持甚至提高Zeigler-Natta催化剂的活性。
1.1.1 单组分载体体系
Rong等[3-5]对坡缕石纳米晶纤维进行高温处理,以防止黏土所含水分对催化剂的毒害作用,然后利用坡缕石纳米晶纤维上的镁离子空穴负载TiCl4催化剂,再以该负载催化剂催化乙烯聚合,得到坡缕石/聚乙烯复合材料。他们考察了聚合反应温度和烷基铝助催化剂类型对该聚合过程的影响,并通过透射电镜(TEM)证明了坡缕石纳米晶纤维均匀分布于聚乙烯基体之中;此外,还发现了聚乙烯的结晶度随着坡缕石纳米晶纤维含量的增加而下降,认为坡缕石纳米晶纤维与聚乙烯基体之间强的界面相互作用导致了复合材料力学性能的提高。
Ramazani等[6]为了提高Zeigler-Natta催化剂的负载量,首先对蒙脱土进行酸处理以增加其羟基含量与孔隙率,然后用烷基铝与酸处理后的蒙脱土反应得到蒙脱土负载铝氧化合物,最后通过MCl4催化剂(M为Ti或V)与蒙脱土负载铝氧化合物反应制备负载催化剂,如图2所示。通过X射线衍射(XRD)和扫描电镜(SEM)表征,发现蒙脱土以剥离或者是插层形式存在于聚乙烯基体之中,且蒙脱土即使在含量较高的情况下依然能够分布均匀;热失重分析(TGA)的结果表明复合材料具有良好的热稳定性,而差示扫描量热(DSC)的测试结果则表明复合材料的结晶度大幅下降;此外,蒙脱土纳米片层的加入使得复合材料的气体阻隔性有了显著提高。

图2 三异丁基铝、四氯化钛、黏土反应原理示意图[6]
Cui等[7]以蒙脱土负载聚氧酸钛盐为催化剂、三异丁基铝为助催化剂制备了蒙脱土/聚乙烯复合材料。TEM实验结果证明了蒙脱土片层在聚乙烯基体中能有效剥离和均匀分布;与纯聚乙烯相比,复合材料的结晶度有所下降,而热稳性与力学性能则明显升高。
Jin等[8]以带有羟基的有机铵盐对黏土进行改性,如图3所示。由于羟基能够有效地将TiCl4催化剂固定在黏土片层之间,因此可得到黏土片层完全剥离的聚乙烯基复合材料,但该催化体系的催化活性明显偏低。

图3 四氯化钛负载于带羟基蒙脱土的反应机理[8]
1.1.2 复合载体体系
Yang等[9]将MgCl2与TiCl4依次负载于黏土片层之间,再原位聚合得到了黏土/聚乙烯复合材料,其原理如图4所示。从图4可见,利用黏土片层间的羟基来连接MgCl2,再通过Mg—Cl—Ti键将催化剂固定在黏土片层之间。XRD和TEM的实验结果表明,黏土片层在乙烯聚合过程中发生层间剥离,以单片层或数片叠层的形式无规地分散于聚乙烯基体之中;与相对分子质量相近的纯聚乙烯相比,极低用量的黏土(质量分数低于1%)就能使复合材料的屈服强度、拉伸强度和拉伸模量有大幅提高。
He等[10]以有机改性黏土-氯化镁(经四氢呋喃处理)双组分载体负载TiCl4催化剂,然后以三乙基铝为助催化剂,通过淤浆聚合得到了有机改性黏土/聚乙烯复合材料。XRD与TEM实验结果证实,有机改性黏土能在聚乙烯基体中有效剥离并均匀分布,有机黏土的加入能够有效地提高复合材料的熔融峰温度、结晶峰温度、热稳定性,但却使其结晶度有所下降。

图4 四氯化钛催化剂插层负载机理[9]
Nikkhah等[11]以膨润土-氯化镁(乙醇盐型)双组分载体负载TiCl4催化剂,然后以三异丁基铝为助催化剂,通过淤浆聚合得到了膨润土/聚乙烯复合材料。研究表明,膨润土已经在聚乙烯基体中完全剥离,且仅仅质量分数1%的膨润土填充量就使得复合材料的氧气阻隔性能比纯聚乙烯提高了200%;虽然复合材料的结晶峰温度、热稳定、杨氏模量、拉伸强度都比纯聚乙烯有所提高,但韧性却有所下降。
Cui等[12]以黏土与氯化镁作为双组分载体的VOCl3负载催化剂制备了完全剥离型的黏土/聚乙烯复合材料。该催化剂通过氯化镁提供负载点,避免了VOCl3与黏土的直接接触,从而具有较高的催化活性。他们发现,黏土片层能有效地剥离;与纯聚乙烯相比,复合材料的熔融峰温度与热稳定性都有所提高,不同黏土含量的黏土/聚乙烯复合材料的力学强度高出了3.4~7.9 MPa,杨氏模量则提升了23.4%~45.3%。
尽管黏土-氯化镁双组分载体能够维持Zeigler-Natta催化体系的高活性,但由于黏土与氯化镁的结合效果较差,不能得到均匀的双组分混合物。而处于游离态的氯化镁在负载Zeigler-Natta催化剂之后,只能催化乙烯聚合得到不含黏土的纯聚乙烯,因此可能会对复合材料的结构造成不良影响。为了克服上述问题,Abedi等[13-15]以丁基辛基镁替代氯化镁与蒙脱土复合,制备了混合均匀的双组分载体,并以该双组分载体负载TiCl4催化剂,然后在助催化剂三乙基铝的作用下催化乙烯聚合,得到了蒙脱土/聚乙烯复合材料,其催化剂负载过程如图5所示。他们考察了影响聚合过程的因素,如聚合反应温度、乙烯气体压强、氢气链转移剂用量、Al/Ni摩尔比等,发现该负载催化剂具有较好的催化活性,且复合材料的热稳性能与力学性能都比纯聚乙烯有所提高,并存在一个性能最佳的蒙脱土用量。

图5 四氯化钛催化剂负载过程中可能发生的反应[14]
1.2 黏土负载茂金属催化剂体系

Huang等[17-19]在有机黏土负载茂金属催化剂方面进行了一系列工作。他们将茂金属催化剂Et[Ind]2ZrCl2负载于有机改性黏土,再以MAO为助催化剂催化乙烯与烯醇共聚得到复合材料,其制备路线如图7所示。由于聚乙烯带有的羟基与剥离的黏土片层之间有着较强的界面相互作用,使得该复合材料即使经过高温处理依然显示出很好的结构稳定性。此外,他们还研究了有机改性黏土类型以及聚合反应溶剂对Et[Ind]2ZrCl2负载催化剂的催化效率和聚乙烯基复合材料结构与性能的影响。
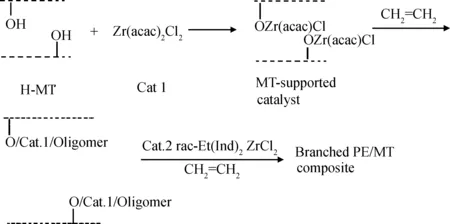
图6 蒙脱土负载双功能催化剂制备蒙脱土/聚乙烯复合材料[16]

图7 通过原位插层共聚方法制备羟基功能化PE/OMMT纳米复合材料[17]
Carrero等[20]以海泡石负载(nBuCp)2ZrCl2催化剂制备了海泡石/线性低密度聚乙烯复合材料,并考察了不同负载方式对催化体系活性的影响。他们发现,所得复合材料的形貌结构较好,而且力学性能也有了较大提高。
Wei等[21]使用二氧化硅改性黏土负载茂金属催化剂Cp2ZrCl2制备得到了黏土-二氧化硅/聚乙烯三元复合材料,制备路线如图8所示。他们通过TEM观察到了剥离的黏土片层与二氧化硅纳米颗粒分散于聚乙烯基体之中,且聚合所得产物具有使用负载催化剂体系所特有的良好颗粒形态和高堆密度。此外,该黏土-二氧化硅/聚乙烯复合材料还显示出较好的力学性能。

图8 原位聚合法制备黏土-二氧化硅/聚乙烯三元复合材料路线[21]
Liu等[22-23]使用多种有机改性剂对黏土进行有机改性,然后分别负载Cp2ZrCl2催化剂来催化乙烯均聚以及乙烯与1-辛烯的共聚,制备路线如图9所示。与Cp2ZrCl2均相催化剂相比,该负载催化剂能够更为有效地调控聚合产物的相对分子质量、化学组成与结构、堆密度等。
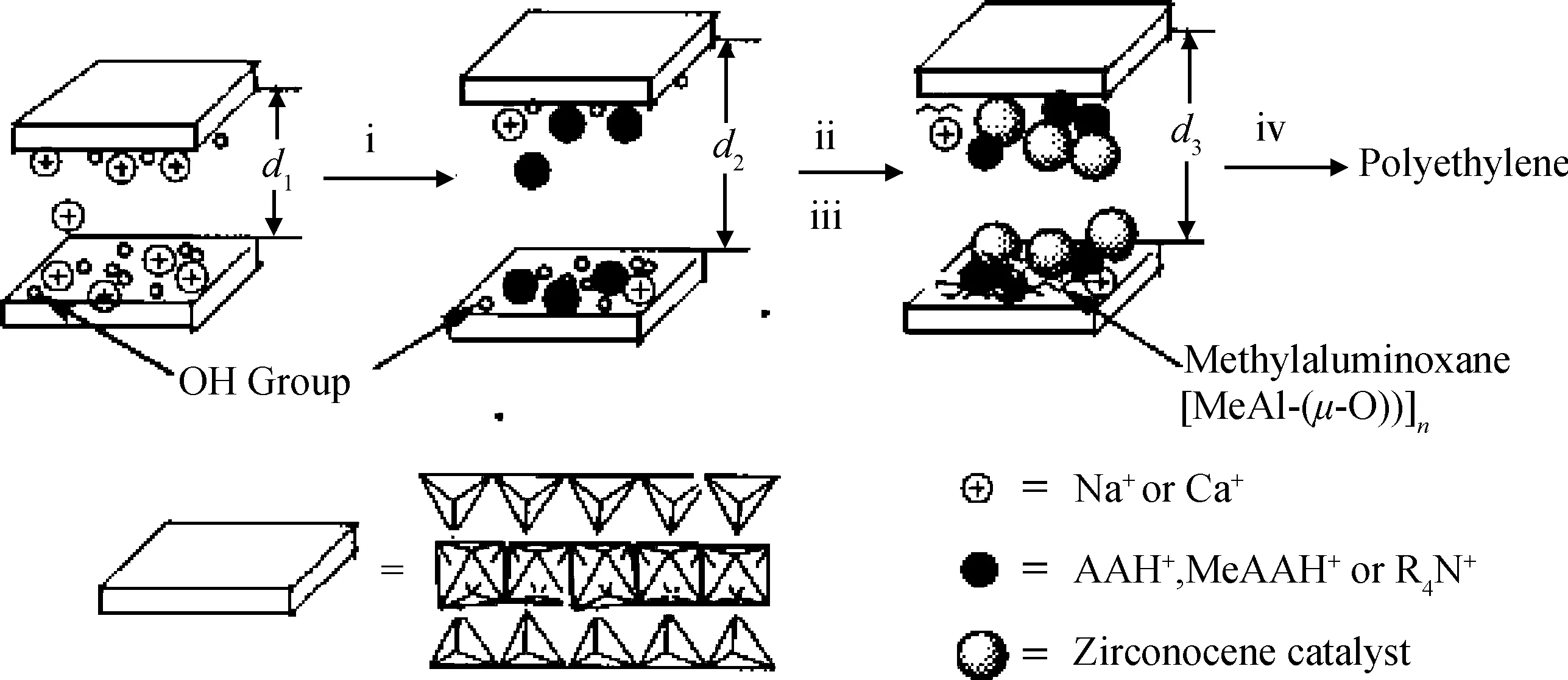
图9 黏土改性、催化剂的负载以及乙烯的聚合示意图[22]
Leone等[24]将MAO负载于有机黏土Cloisite 15A,并将其作为Cp2ZrCl2催化剂的助催化剂催化乙烯聚合。该体系的催化活性要高于均相催化剂体系,并且随着MAO含量的增加而增加;采用元素分析、TGA、TGA-傅里叶转换红外光谱(FTIR)、XRD、TEM等手段对被MAO负载的有机黏土表征后发现,MAO与黏土片层之间的有机改性剂发生了反应,破坏了黏土片层的有序结构。他们还对所得复合材料的形貌和热行为进行了分析研究。
Xalter等[25]将MAO负载于有机改性勃姆石,并将其作为Cp2ZrCl2催化剂的助催化剂催化乙烯聚合。他们发现,以未改性勃姆石和含2%有机改性剂的勃姆石为载体时,催化体系的活性较高,但勃姆石在聚乙烯基体中的分散效果较差;以含20%有机改性剂的勃姆石为载体时,催化体系的活性较低,但勃姆石的分散效果较好。
Maneshi等[26]以三甲基铝处理不同类型的有机改性黏土后,将其分别作为Cp2ZrCl2催化剂的载体制备负载催化剂,如图10所示,并研究了该负载催化剂的催化活性以及所得复合材料的形貌。他们发现,有机改性剂的类型对于催化剂的负载效果有着非常重要的影响。由叔胺盐改性的有机黏土Cloisite 93A具有非常好的催化活性,而其它由季胺盐改性的有机黏土则催化活性较差,甚至完全没有活性。

图10 茂金属催化剂负载于蒙脱土的可能机理[26]
1.3 黏土负载后过渡金属催化剂体系
作为早期开创性的工作之一,Heinemann等[27]采用熔融共混和原位聚合两种方法制备了黏土/高密度聚乙烯复合材料和黏土/线性低密度聚乙烯复合材料,发现使用后过渡金属催化剂或者茂金属催化剂进行原位聚合,较之熔融共混方法更有利于黏土片层的剥离分散;通过原位聚合法得到的黏土/聚乙烯复合材料其刚性、强度、韧性、耐热变形性和阻隔性能都有显著提高,且材料透明性没有下降。因此,他们认为该材料具有非常大的应用前景。
He等[28-30]以经过三乙基铝处理的有机黏土作为载体制备镍系后过渡金属负载催化剂,如图11所示,并研究了Al/Ni摩尔比、聚合反应时间、聚合反应温度等因素对该负载催化剂聚合催化活性和复合材料黏土填充量的影响。TGA的结果表明,有机黏土的加入能够明显地提高该复合材料的热稳定性。然后,进一步利用了多面齐聚倍半硅氧烷(POSS)的可反应性,通过阳离子交换反应,对无机黏土进行改性,并以该改性黏土作为镍系后过渡金属催化剂的载体,再通过原位聚合制备了黏土与POSS协同增强的聚乙烯基复合材料。由于在负载过程中催化剂通过共价键固定在黏土的片层之间,因此在聚合反应开始之后,随着聚乙烯分子链在黏土片层之间的不断增长,黏土片层最终能够完全剥离,并均匀分布于聚乙烯基体之中。核磁共振碳谱(13C-NMR)的研究结果表明,将该催化剂负载到黏土片层之间后,由于黏土片层纳米空间结构的影响,在聚合过程中镍系后过渡金属催化剂的链行走行为受到了很大限制,从而改变了聚乙烯产物的支链分布情况,并显著降低了聚乙烯产物的支化度,导致复合材料的熔点和结晶度均高于纯聚乙烯。此外,黏土片层对催化剂的空间效应作用不仅降低了聚合反应的链行走速率,还同时降低了链转移和链终止反应的速率,从而增大了聚乙烯产物的相对分子质量。进一步使用连续自成核/退火方法分析了纯聚乙烯以及POSS改性黏土/聚乙烯复合材料的聚乙烯基体,结果表明,负载催化剂与均相催化剂所得聚乙烯产物的支化非均匀性存在明显差异;POSS改性黏土/聚乙烯复合材料的热稳定性要高于纯聚乙烯,当复合材料中POSS改性黏土含量为质量分数9.75%时,其50%的热失重温度要比纯聚乙烯高62℃。动态机械热分析(DMA)的研究结果表明,与纯聚乙烯相比,POSS改性黏土/聚乙烯复合材料的储存模量有了大幅提高,当POSS改性黏土质量分数为9.75%时,其在20℃的储存模量提高了1226%。

图11 通过原位聚合制备黏土与POSS协同增强的聚乙烯基复合材料[29]
Shin[31]通过多步反应,如图12所示,将乙烯基反应基团连接到黏土片层之间,然后再负载镍系化合物得到后过渡金属负载催化剂。在聚合过程中,乙烯单体进入到黏土片层之间进行聚合,使得黏土片层有效剥离,并且生成的聚乙烯分子链通过共价键与黏土片层相连,增加了黏土与聚乙烯基体之间的界面相互作用。
Ray等[32]将铁系后过渡金属催化剂负载于经过MAO预先处理的蒙脱土后得到负载催化剂,并将其用于催化乙烯聚合。他们发现,与以未经MAO处理蒙脱土作为催化剂载体的体系相比,该方法能更有效地使黏土片层剥离;负载催化剂的活性与Al/Fe摩尔比无关,而聚乙烯基体的结晶度和晶粒尺寸则会受到蒙脱土剥离程度的影响。
Mignoni等[33]以有机改性蒙脱土为载体,通过镍系后过渡金属-MAO(或三甲基铝)体系催化乙烯聚合,结果表明,催化剂活性与黏土填充量可以通过改变Al/Ni摩尔比等因素进行有效调控;黏土含量的改变对复合材料的性能有着明显的影响;由于蒙脱土纳米片层有效剥离并均匀分布于聚乙烯基体之中,提高了复合材料的力学性能、结晶峰温度、熔融峰温度。

图12 通过双功能有机改性剂对黏土进行改性并原位聚合制备共价键连接的黏土/聚乙烯复合材料[31]
通常,对黏土进行有机改性用到的都是有机小分子,有可能对催化剂活性和复合材料性能造成不良影响。Scott等[34]针对这一问题,以经过酸处理的蒙脱土负载镍系后过渡金属催化剂催化乙烯聚合反应,如图13所示。他们认为,催化剂配体上的亚甲基和蒙脱土片层上的铝元素存在着路易斯酸碱相互作用,可以在不使用有机改性的情况下,使催化剂直接负载到黏土的片层之间。实验结果表明,该催化体系的活性较高,并且能够实现黏土片层的有效剥离。
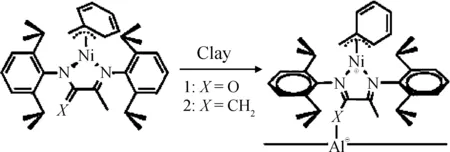
图13 黏土活化镍系后过渡金属催化剂的可能机理[34]
Leone等[35]将铁系后过渡金属催化剂负载于经MAO改性的黏土后得到负载催化剂,并将其催化乙烯聚合得到复合材料。他们发现,(1)由于催化剂被负载之后活性中心的分散性较好,表现出比均相催化剂更高的活性;(2)由于黏土载体的保护作用,负载催化剂比均相催化剂具有更长的反应寿命;(3)由于黏土载体能够降低链转移的速率,使得产物的相对分子质量增加。
2 碳纳米管/聚乙烯复合材料
碳纳米管由于其独特的纳米结构和优良的电、磁、光、力学和热传导等性能,引起了人们广泛重视。将碳纳米管作为填料与聚合物共混,对提高聚合物性能以及实现材料的功能化具有重要意义。然而,由于碳纳米管之间存在范德华力作用并且极易缠绕团聚,要将碳纳米管应用在聚合物基体中并充分发挥其优异性能,具有相当的难度[93]。制备碳纳米管复合材料的两种主要方法是共混法和原位聚合法。与共混法相比,原位聚合法获得的复合材料中碳纳米管分散更均匀,并且碳纳米管与聚合物基体之间具有更强的相互作用[36]。
2.1 碳纳米管负载Zeigler-Natta催化剂体系
Ramazani等[37-38]采用以二乙氧基镁和带羟基多壁碳纳米管作为双组分载体的TiCl4负载催化剂,制备了多壁碳纳米管/超高分子量聚乙烯,其催化剂负载机理如图14所示。研究结果表明,碳纳米管在复合材料中的分散效果良好;碳纳米管能够明显提高复合材料的杨氏模量、屈服应力和断裂拉伸强度。他们还采用TGA表征了该复合材料的热稳定性,并采用Friedman、Ozawa、Flynn、Wall和Kissinger等动力学方法研究了复合材料的热失重行为,发现碳纳米管的加入能够增加材料的热分解活化能,从而提高了复合材料的热稳定性。
Park等[39]采用TiCl4-EtAl3催化体系制备了碳纳米管/超高分子量聚乙烯复合材料。摩擦实验的结果表明,与纯超高分子量聚乙烯和熔融共混制备的碳纳米管/超高分子量聚乙烯复合材料相比,原位聚合制备的碳纳米管/超高分子量聚乙烯复合材料具有更好的耐磨性。他们认为其原因主要是,(1)原位聚合制备的碳纳米管/超高分子量聚乙烯复合材料其碳纳米分散均匀,与聚乙烯基体界面作用力较强;(2)原位聚合制备的碳纳米管/超高分子量聚乙烯复合材料其热传导系数更高,能够及时地将摩擦过程中产生的热量传递出去,降低摩擦表面的温度,减少了摩擦的损耗。
Liu等[40]先用格氏试剂处理带羧基、羟基等功能基团的多壁碳纳米管,然后负载TiCl4催化剂,再以AlEt3为助催化剂催化乙烯聚合,如图15所示,制备得到了具有“芯-皮”结构的多壁碳纳米管/聚乙烯复合材料,并研究了该材料的结晶行为、热稳定性、介电性能。

图14 以二乙氧基镁和带羟基多壁碳纳米管负载四氯化钛催化剂的机理[37]

图15 多壁碳纳米管/聚乙烯复合材料制备过程示意图[40
2.2 碳纳米管负载茂金属催化剂体系
茂金属负载催化剂体系是通过原位聚合制备碳纳米管/聚乙烯复合材料的最常用催化体系。通过该体系制备复合材料的一般过程如图16所示[41-43]。其中步骤(i)与步骤(ii)不是必须步骤。碳纳米管表面经过处理后引入了羧基、羟基等功能基团得到功能化碳纳米管,功能基团的引入有利于MAO和催化剂的负载;将MAO负载于功能化碳纳米管表面,可以避免杂质对催化剂的毒害作用;在碳纳米管上负载催化剂,在助催化剂作用下,催化乙烯聚合得到复合材料。某些碳纳米管负载茂金属催化剂体系甚至可以表现出比均相催化剂更高的活性[42-43],其原因可能是催化剂在负载后分散得更好,不像均相催化剂那样团聚在一起,从而能够使得催化剂活性中心与乙烯单体小分子充分接触,并避免了双分子失活现象。

图16 茂金属催化剂负载过程以及其催化乙烯聚合示意图[41]
Tang等[44]首先经过多步反应将乙烯基团接枝到多壁碳纳米管表面,然后在rac-(en)(THInd)2ZrCl2-MAO催化体系作用下,催化改性碳纳米管与乙烯共聚合得到多壁碳纳米管/聚乙烯复合材料,如图17所示。1H-NMR、SEM、FTIR、TGA等表征结果证明了聚乙烯成功地接枝到了碳纳米管表面。

图17 多壁碳纳米管/聚乙烯复合材料制备过程示意图[44]
Kim等[45-47]则采用一锅法制备了多壁碳纳米管/聚乙烯复合材料。先依次用三异丁基铝与MAO对甲苯溶剂中的多壁碳纳米管进行处理,然后直接向溶液中加入rac-Et(Ind)2ZrCl2催化剂,再通入乙烯单体进行反应,得到复合材料。他们发现,碳纳米管的加入能够有效地提高复合材料的热传导系数、电导率、复合黏度、储存模量、损耗模量、冲击强度等性能。他们还进一步研究了该复合材料的热稳定性和非等温结晶动力学过程。
Bahuleyan等[48]采用了一种相对比较简单的一锅法制备了多壁碳纳米管/聚乙烯复合材料。首先在甲苯溶剂中加入Cp2TiCl2催化剂(或Cp2ZrCl2催化剂)和碳纳米管,然后对其进行超声波处理,最后加入助催化剂并通入乙烯气体进行聚合反应。他们发现,通过选择不同类型的催化剂和聚合反应条件,可以控制复合材料的外观形貌,得到类似于“香肠”形貌或者是串晶形貌的复合材料。
Dong等[49-50]以经MAO处理的多壁碳纳米管作为载体负载Cp2ZrCl2催化剂催化乙烯聚合。在该聚合过程中(T=50℃,[Al]/[Zr]=1000),生成的聚乙烯分子链以高长/径比的碳纳米管作为模板不断进行包裹,最终得到了纤维状形貌的复合材料;当[Al]/[Zr]摩尔比和聚合温度继续升高时,纤维状形貌将向无定型形貌转变。他们进一步发现,分别以末端封闭碳纳米和末端开口碳纳米管作为载体时,可以分别得到纤维状和无定型两种不同的聚乙烯形貌。他们认为,以开口碳纳米管作为载体时,催化剂可以进入碳纳米管的内部,聚合反应开始之后随着聚乙烯分子链在碳纳米管内部不断地增长,最终导致碳纳米分裂成为碎片;后续生成的聚乙烯将以不规则的碳纳米管碎片作为模板不断包裹,从而得到了具有不规则形貌的聚乙烯,其机理如图18所示。Trujillo等[51]以类似的方法分别制备了以单壁碳纳米管、双壁碳纳米管、多壁碳纳米管为填料的聚乙烯基复合材料,测定了反应终止后助催化剂的残留物三氧化二铝在复合材料中的含量,同时还考察了复合材料的形貌结构、晶体成核过程、结晶行为等,发现各种类型的碳纳米管对聚乙烯都有着很好的结晶成核作用。
Toti等[52]用改性甲基铝氧烷(MMAO)对多壁碳纳米管进行预处理,然后负载双组分催化剂,得到催化剂体系1 CoCl2N2Th+[(η5-C5Me4)SiMe2(NtBu)]-TiCl2和催化剂体系2 CoCl2N2Th+Cp2ZrCl2,前者为乙烯齐聚催化剂,后者为乙烯共聚催化剂,再催化单一乙烯单体进行聚合,制备了多壁碳纳米管/线性低密度聚乙烯复合材料;通过改变催化剂组分的比例和共聚合反应的条件可以有效地控制聚乙烯基体的支化度以及接枝链类型。
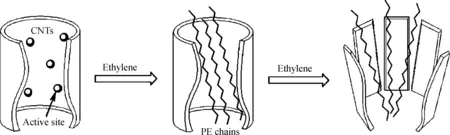
图18 Dong等所制备的无规聚乙烯的形成过程[49]
Park等[53-54]采用一锅法制备了多壁碳纳米管/高密度聚乙烯复合材料。在反应釜中依次加入正庚烷、MAO、多壁碳纳米管与Cp2ZrCl2催化剂的正庚烷淤浆混合物后,再通入乙烯气体进行聚合反应。力学测试的结果表明,多壁碳纳米管的加入使得复合材料的杨氏模量与纯聚乙烯相比提高了359%。他们进一步的研究发现,碳纳米管可以与带正电的二茂锆活性中心形成配位键,并充当具有强给电子能力的配体;钛的亲电活性可以使得碳纳米管和半茂金属钛(Cp*TiCl3,Cp*:五甲基环戊二烯基)有强烈的电子相互作用,从而影响附着在碳纳米管表面的半茂钛催化剂的催化性能;Cp*TiCl3可以通过Cp*与多壁碳纳米管表面的相互作用从而吸附于碳纳米管壁,形成碳纳米管-Cp*TiCl3负载催化剂;以该催化体系催化乙烯聚合得到的复合材料,其多壁碳纳米被超高分子量聚乙烯包裹后直径为30~70 nm,大于碳纳米管直径10~15 nm。

图19 Park等制备的无规聚乙烯的形成过程[54]
2.3 碳纳米管负载后过渡金属催化剂体系
采用后过渡金属催化剂体系制备碳纳米管/聚乙烯复合材料的报道相对较少。Zhang等[55]制备了含有大体积配体的高活性铁系后过渡金属催化剂,如图20所示。该铁系催化剂能够通过π-π相互作用负载到多壁碳纳米管的表面,且催化活性要好于均相催化剂。研究表明,多壁碳纳米管能够均匀分散在聚乙烯基体之中。

图20 铁系后过渡金属催化剂负载于多壁碳纳米管的示意图[55]
2.4 母料共混体系
原位聚合法的优点在于碳纳米管分散比较均匀,与聚乙烯基体之间的界面作用较强,且能够制备高填充碳纳米管/聚乙烯复合材料。因此不少科研工作者以含有高碳纳米管填充量的聚乙烯基复合材料作为母料与聚合物熔融共混制备相应的复合材料。
何富安[56]采用母料熔融共混(mPEC)与直接熔融共混(dPEC)2种方法制备了多壁碳纳米管/聚乙烯复合材料。在mPEC制备过程中,首先通过TiCl4-三乙基铝催化体系原位聚合制备多壁碳纳米管填充量为质量分数53%的聚乙烯基复合材料,如图21所示,然后以其作为母料与高密度聚乙烯树脂熔融共混。研究结果表明,mPEC的力学性能要优于dPEC,说明在原位聚合过程中生成的聚乙烯包裹层对多壁碳纳米管的分散以及界面改性起到了非常重要的作用;多壁碳纳米管质量分数为3%的dPEC3和mPEC3的拉伸屈服强度分别为25.23 MPa和27.86 MPa,比纯聚乙烯提高了25.2%和38.3%,杨氏模量分别为963 MPa和1081 MPa,比纯聚乙烯提高了26.2%和41.7%;碳纳米管质量分数为1%的dPEC1和mPEC1的弯曲强度为29.50 MPa和31.55 MPa,分别比纯聚乙烯提高了16.3%和24.4%,弯曲模量为753 MPa和883 MPa,分别比纯聚乙烯提高了21.8%和42.9%。与之相类似,Tong[36]等的研究结果则表明,采用母料熔融共混法得到的单壁碳纳米管(质量分数1%)/聚乙烯复合材料的屈服强度、拉伸强度、拉伸模量、断裂伸长率、断裂能分别比直接熔融共混法得到的复合材料提高了25%、15.2%、25.4%、21%、38%。此外,Vega等[57-58]先以多壁碳纳米管作为载体负载Cp2ZrCl2催化剂,然后催化乙烯聚合得到母料,再与高密度聚乙烯熔融共混得到复合材料,并研究了该复合材料的熔融流变行为、结晶行为、力学性能等。
Bredeau等[59]采用以碳纳米管负载rac-Et(Ind)2ZrCl2-MAO催化剂体系催化乙烯与降冰片烯共聚,得到共聚物基复合材料,并以该共聚物基复合材料作为母料采用熔融共混法制备了碳纳米管/乙烯-醋酸乙烯共聚物复合材料。力学性能测试表明,碳纳米管的加入能够大幅度提高复合材料的杨氏模量。此外,他们还在碳纳米管上依次负载MMAO和Cp2*TiCl2催化剂,然后催化乙烯聚合制备得到母料,再与乙烯-醋酸乙烯共聚物熔融共混得到复合材料。他们的研究表明,母料熔融共混法可以比直接熔融共混法得到更好的碳纳米管分散效果,从而有助于提高复合材料的热稳定性、力学性能、导电性能。
为了避免由于使用MAO、烷基铝等助催化剂而导致聚乙烯产物中含有大量的氧化铝残余物,Ravasio等[60]以[Sc(g5-C5Me4SiMe3)(g1-CH2SiMe3)2(THF)]作为催化剂,[Ph3C][B(C6F5)4]为助催化剂,少量三异丁基铝作为清除剂,制备了多壁碳纳米管/聚乙烯-降冰片烯共聚物复合材料,如图22所示。他们发现,在以该复合材料为母料与TOPAS环烯烃类共聚物进行熔融共混得到的复合材料中,碳纳米管分散均匀。

图21 多壁碳纳米管/聚乙烯母料的制备过程[56]

图22 多壁碳纳米管/聚乙烯-降冰片烯共聚物复合材料母料的制备过程[60]
3 石墨类填料/聚乙烯复合材料
石墨类填料包括普通石墨、氧化石墨、纳米石墨片、石墨烯等。其中的石墨烯是近几年继富勒烯、碳纳米管后发展起来的又一种新型碳纳米材料,在化学、物理、材料、电子等领域都掀起了研究热潮。石墨烯具有优异的导电性和传热性、超高的模量、高的长/径比(长度和厚度的比值)和极大的比表面积,是理想的增强剂。
Alexandre等[61-62]采用3种不同方法制备了石墨/聚乙烯复合材料。一种方法是将催化剂负载于经过MAO改性的石墨,然后进行原位聚合;另一种方法是一锅法,即将石墨加入溶剂中,再依次添加助催化剂MAO与催化剂进行原位聚合;第3种方法即是机械共混法。他们还研究了不同方法制备得到的石墨/聚乙烯复合材料的外貌形态、热性能、导电性能等。
Fabiana等[63-64]以Cp2ZrCl2为催化剂,MAO为助催化剂,纳米石墨片为填料,制备了纳米石墨片/聚乙烯复合材料。在催化过程中,聚乙烯分子链不断对其插层,使得石墨纳米片层不断剥离,并掺杂进聚乙烯基体均匀分散,由于纳米石墨片的存在降低了Cp2ZrCl2催化剂的活性。他们进一步发现,纳米石墨片的加入能够使复合材料的热失重起始温度和剥离温度分别提高30℃和5℃,并且存储模量和电导率也有所上升。
Hu等[65]用经过有机硅氧烷处理的氧化石墨烯先后负载MAO与Cp2ZrCl2,并用该负载催化体系制备了聚乙烯基复合材料。由于有机基团被引入到石墨烯片层之间,使得石墨烯片层在聚合过程中能够有效剥离,并均匀分散在聚乙烯基体之中,其制备过程如图23所示。使用非有机改性氧化石墨烯负载催化剂得到的复合材料则出现了明显的相分离。此外,氧化石墨烯/聚乙烯复合材料的熔点比采用均相催化剂得到的纯聚乙烯有明显提高。
Lee等[66]在多层石墨烯上负载了(n-BuCp)2ZrCl2催化剂后,在助催化剂MAO的作用下催化乙烯聚合物,同样发现了聚乙烯分子链对多层石墨烯的插层剥离现象,而且石墨烯在聚合过程中起到了形貌模板的作用。石墨烯负载(n-BuCp)2ZrCl2催化剂的过程如图24所示。TEM的结果表明,生成的复合材料具有类似于菊花花瓣的形貌。此外,石墨烯还起到了催化剂大配体的作用,使得到的聚乙烯比均相催化剂具有更高的相对分子质量和多分散系数。

图23 茂金属催化剂负载于氧化石墨烯的过程[65]
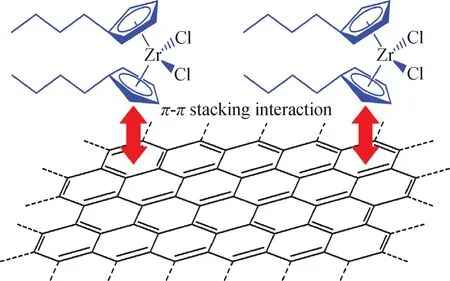
图24 (n-BuCp)2ZrCl2通过π-π作用负载于多层石墨烯的示意图[66]
Todd等[67]在较低温度(40℃)和压力(乙烯气体压强为1.01×105Pa)下,以热还原石墨烯-(n-BuCp)2ZrCl2)-MAO催化体系制备了聚乙烯基复合材料。结果表明,当热还原石墨烯质量分数为5.2%时,复合材料的拉伸强度和杨氏模量比纯聚乙烯分别提高了57%和170%。
Stürzel等[68]以带羟基的功能化石墨烯为载体,采用铬系催化剂和MAO助催化剂在高压反应釜中原位制备了石墨烯/超高分子量聚乙烯复合材料,并与其它碳系填料负载催化剂体系以及勃姆石负载催化剂作比较,负载过程如图25所示。由于石墨烯带有的羟基能够很好地使负载催化剂分散在正庚烷中,因此该催化体系表现出最好的催化活性和形貌控制能力;当功能化石墨烯的质量分数仅为1%时,就能够大幅度提高复合材料的刚性、断裂伸长率和结晶能力。

图25 铬系催化剂负载于功能石墨烯的过程[68]
Choi等[69]则以氮掺杂还原氧化石墨烯作为Cp2ZrCl2与Cp2TiCl2催化剂的载体,制备多壁碳纳米管/超高分子量聚乙烯复合材料。他们发现,在聚合物过程中氮掺杂石墨烯起到了相当于配体的作用,能够调节金属中心空间和电子属性,所以得到的聚乙烯比使用均相催化剂得到的聚乙烯具有更高的相对分子质量,其制备过程如图26所示。
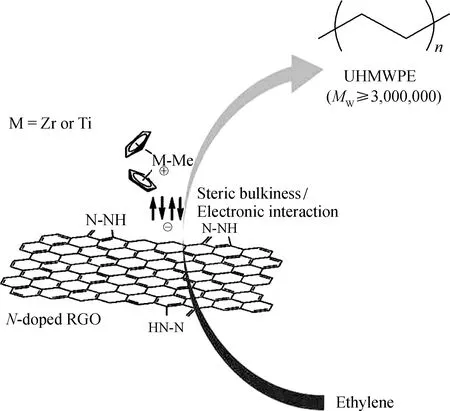
图26 茂金属催化剂负载于氮掺杂还原氧化石墨烯以及制备多壁碳纳米管/超高分子量聚乙烯复合材料[69]
4 原位聚合制备其它聚乙烯基复合材料
除了上述3种主要填料之外,科研工作者还将原位聚合法应用于制备其它聚乙烯基复合材料,在提高聚乙烯材料的性能以及实现聚乙烯功能化方面都取得了相当的成果。
作为早期开创性的工作之一,Alexandre[70-71]等使用了不同的催化剂载体,包括玻璃珠、石墨、氢氧化镁、高岭土等等,负载茂金属催化剂,并通过原位聚合制备了多种无机填料/聚乙烯复合材料。
He等[72-73]以经过有机改性的水滑石作为载体负载镍系后过渡金属催化剂后,再通过原位聚合法制备了水滑石/聚乙烯复合材料,如图27所示。考察了各种聚合条件,包括聚合反应时间、聚合反应温度和Al/Ni摩尔比对负载催化剂活性的影响,结果表明,(1)随着反应时间的延长,负载催化剂的活性逐渐下降;(2)随着聚合温度的升高,负载催化剂催化活性先增大后减小,在30~40℃范围内达到最大值;(3)随着Al/Ni摩尔比的增大,负载催化剂的活性也增大,Al/Ni摩尔比在300~900之间时,活性的增长速率较快,当Al/Ni摩尔比达到1200后催化剂活性增长的速率放缓。他们进一步研究了负载催化剂与非负载均相催化剂在相同聚合条件下的聚合反应动力学,结果表明,对催化剂进行负载后,提高了催化剂的活性中心的长期稳定性;在负载催化剂体系中,水滑石对生成的聚乙烯具有很好的模板作用,得到了颗粒状外貌的聚乙烯产物,而使用均相催化剂得到的则是无定型结构聚乙烯;水滑石片层对于负载在片层之间的催化剂存在着受限空间效应,它降低了链转移和链终止反应的速率,从而增大了聚乙烯产物的相对分子质量;水滑石片层以纳米级的尺寸均匀分散于聚乙烯的基体之中,得到了部分剥离型的水滑石/聚乙烯复合材料。由于聚乙烯与水滑石纳米片层之间有着较强的界面作用,从而限制了聚乙烯分子链的运动,并减弱了聚乙烯的结晶能力,因此随着水滑石含量的增加,聚乙烯的结晶度下降;水滑石/聚乙烯复合材料的热稳定性要高于纯聚乙烯,当水滑石质量分数为10.32%时,其30%的热失重温度要比纯聚乙烯高61℃。此外,水滑石/聚乙烯复合材料在熔融状态下的储能模量、损耗模量、复合黏度均高于纯聚乙烯。

图27 镍系后过渡金属催化剂负载于有机改性水滑石以及制备水滑石/聚乙烯复合材料[73]
Monteil等[74]为了增加二氧化硅纳米粒子与聚乙烯基体之间的相容性,首先在二氧化硅纳米粒子表面接枝辛烯基团或者是辛基硅氧烷,然后通过镍系后过渡金属催化剂原位聚合,得到二氧化硅/聚乙烯复合材料。他们发现,通过改变镍系催化剂的配体可以有效地调控二氧化硅的分布形态。
Wang等[75]将Cp2ZrCl2催化剂在MAO的作用下负载于高岭土,如图28所示,然后通过原位聚合得到了高岭土/聚乙烯复合材料。溶剂抽提实验和FTIR的结果显示,聚乙烯基体与高岭土粒子之间存在着较强的界面相互作用;DMA的结果则显示,这种界面作用能够有效地阻碍聚乙烯高分子链的运动。此外,与机械共混相比,原位聚合使得高岭土具有更好的分散性。
Zhang等[76]将Ziegler-Natta催化剂负载于纳米铁电材料(Pb,Sr)TiO3(PST)粉末上,然后再通过控制聚合反应时间,得到不同PST含量的PST/聚乙烯复合材料。结果表明,PST能够均匀地分散于聚乙烯基体之中,并且PST含量对PST/聚乙烯复合材料的介电常数有着很大影响。
Jongsomjit等[77-80]在原位聚合制备线性低密度聚乙烯基复合材料方面进行了大量工作。他们通过二氧化硅和二氧化锆纳米粒子负载rac-Et(Ind)2ZrCl2-MAO催化剂体系催化乙烯与α-烯烃共聚,分别得到了二氧化硅/线性低密度聚乙烯复合材料和二氧化锆/线性低密度聚乙烯复合材料。当使用二氧化硅作为载体时,催化剂的活性较低,但当使用二氧化锆作为载体时,催化剂活性却有大幅度提高,为二氧化硅负载体系的5倍。他们认为,这可能是由于二氧化硅纳米粒子与MAO之间较强的结合力造成了空间位阻,使得乙烯单体难以接近催化剂活性中心。根据TEM的结果,可以看到二氧化硅与二氧化锆纳米粒子在线性低密度聚乙烯基体中都有较好的分散效果。他们还比较了不同粒径(10 nm和15 nm)的二氧化硅作为载体时对催化剂活性的影响,发现当二氧化硅粒径较大时,与MAO之间的相互作用较弱,催化剂具有更高的活性,并且使得α-烯烃接触催化剂活性中心的几率增加,如图29所示,从而导致聚乙烯的支化度上升和熔点下降。此外,他们还通过类似的原位聚合方法制备了三氧化二铝/线性低密度聚乙烯复合材料、氧化锌/线性低密度聚乙烯复合材料和二氧化钛/线性低密度聚乙烯复合材料,并研究了它们的结构与性能[81-83]。

图28 Cp2ZrCl2/MAO负载于高岭土表面形成活性中心[75]
Cheng等[84]通过母料熔融共混法制备了二氧化硅/聚乙烯复合材料。结果表明,母料熔融共混得到的复合材料中二氧化硅的分散效果要比直接熔融共混得到的复合材料更好;在二氧化硅质量分数为2.0%时,母料熔融共混得到的复合材料的最高热失重速率温度、结晶度和拉伸强度分别为472℃、68.3%和28.3 MPa,而直接熔融共混得到的复合材料其相应的数值则为430℃,66.3%和26.3 MPa。
Covarrubias等[85]采用原位聚合和熔融共混两种方法制备了多孔层状磷酸铝/聚乙烯复合材料。他们发现,尽管原位聚合法可以使得多孔层状磷酸铝充分剥离,但其结构却遭到了一定程度的破坏,而熔融共混则能够较好地保持多孔层状磷酸铝的结构。小分子气体H2/CO的渗透测试结果则表明,采用熔融共混得到的多孔层状磷酸铝/聚乙烯复合材料的透气性能要好于采用原位聚合法得到的复合材料。
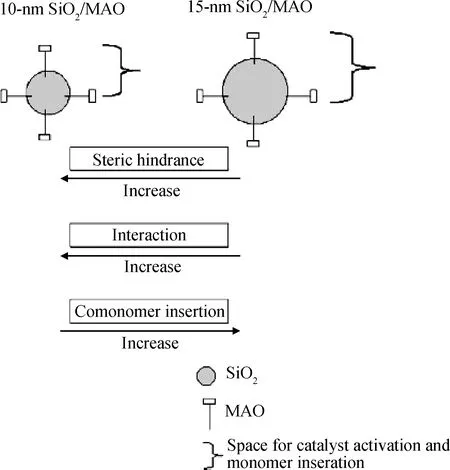
图29 不同尺寸的纳米SiO2对乙烯聚合的影响[77]
Wang等[86]以MCM-41分子筛与MgCl2为复合载体,TiCl4为催化剂制备了MCM-41/聚乙烯母料,然后与聚乙烯树脂熔融共混,得到了相应的复合材料。与纯聚乙烯相比,所得MCM-41(质量分数1%)/聚乙烯复合材料的力学性能有了大幅提高,其拉伸模量、拉伸强度和断裂伸长率分别提高了24.7%、28.3%、23.5%。
Kaleel等[87]以Cp2ZrCl2和Cp2TiCl2为催化剂、MAO为助催化剂、质量分数1%锰掺杂的二氧化钛为填料制备了相应的聚乙烯基复合材料,考察了填料含量、聚合反应温度、乙烯气体压强等因素对复合材料热性能与力学性能的影响。结果表明,使用Cp2ZrCl2催化剂对聚乙烯的熔点影响不大,并能使复合材料的断裂强度、杨氏模量、断裂伸长率有明显提高。
Wang等[88-90]将TiCl4催化剂负载在磁性纳米粒子表面制备得到了磁性纳米催化剂,然后通过催化乙烯原位聚合得到新型磁性聚乙烯复合材料,其制备路线如图30所示。他们研究了该负载催化剂的催化活性,同时还使用不同烷基铝对磁性粒子进行改性,发现三乙基铝的改性效果最好。江洪流等[91]则用纳米四氧化三铁磁性粉末作为载体,负载α-二亚胺镍催化剂制备得到磁性纳米催化剂,通过乙烯在纳米催化剂粒子表面原位聚合得到新型磁性支化聚乙烯复合材料,结果表明,四氧化三铁较均匀地分散在聚乙烯基体中,其尺寸在20 nm左右。
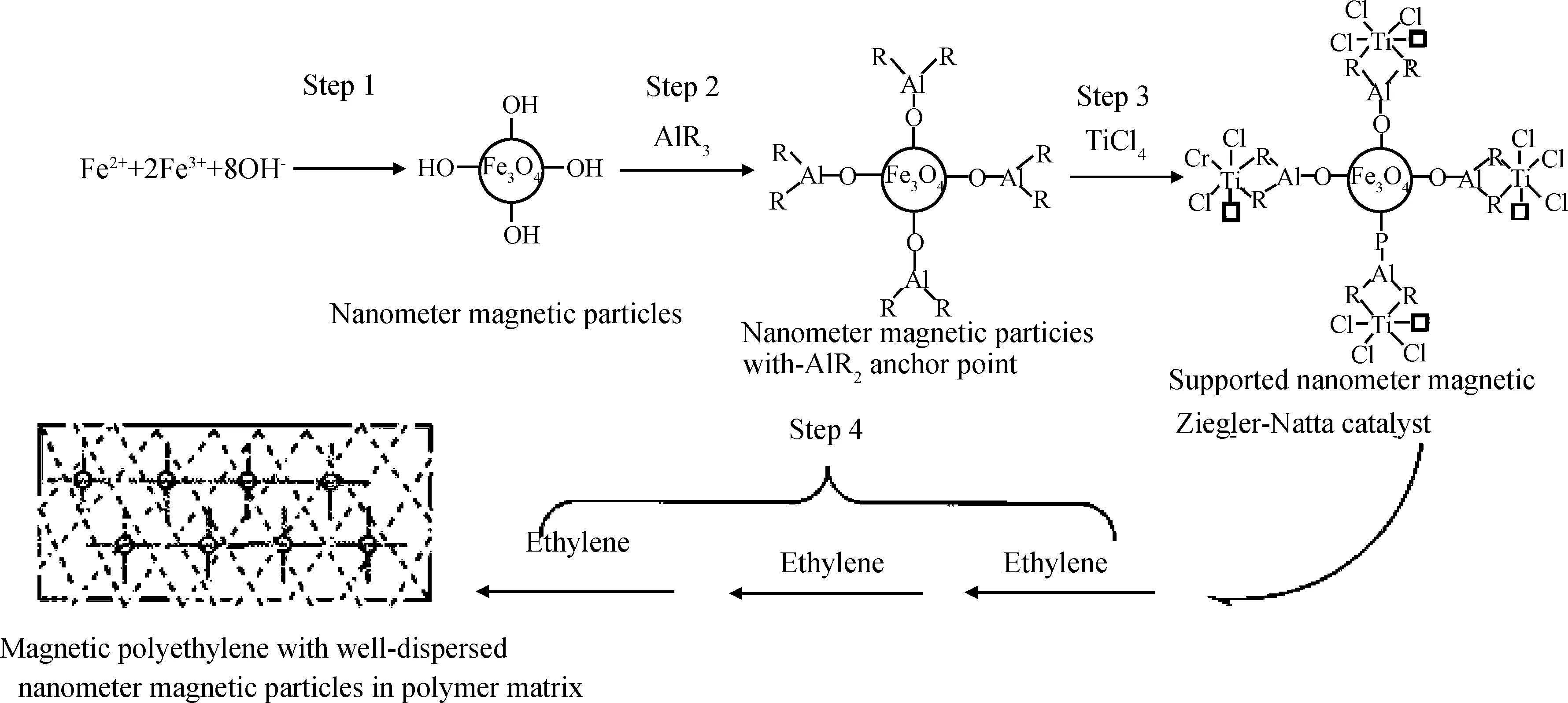
图30 磁性催化剂与磁性聚乙烯复合材料的制备路线[88]
Park等[92]分别采用机械共混和二氧化锆负载TiCl4催化剂催化乙烯原位聚合2种方法制备了二氧化锆/超高分子量聚乙烯复合材料。采用原位聚合方法得到的复合材料的二氧化锆纳米粒子分散得更为均匀,并且与聚乙烯基体有着更强的界面作用,因此力学性能与耐磨性都比使用机械共混方法得到的复合材料有了显著提高。
5 结束语
原位聚合是迄今用于制备填料/聚乙烯复合材料的最有效方法之一,该方法为提高聚乙烯材料的性能以及实现其功能化提供了重要途径。然而,目前该领域研究仍面临以下一些挑战:
(1) 如何使负载催化剂具有与均相催化剂相当、甚至更高的催化活性;
(2) 如何使聚合后得到的聚乙烯基复合材料粉末具有好的颗粒形貌和高的堆密度;
(3) 如何使填料能够在聚乙烯基体中稳定分散,不至于复合材料在加工与使用过程中出现相分离现象;
(4) 如何提高聚乙烯基复合材料的综合性能以及有效地实现其功能化。
因此,深入研究聚乙烯基复合材料的原位聚合反应规律及其影响因素,同时探索有关催化剂效率、产物形貌控制、复合材料结构与性能之间的相互关系,对于高性能聚乙烯基复合材料的研究和工业化生产将具有十分重要的意义。
[1] RAY S S, OKAMOTO M. Polymer/layered silicate nanocomposites: A review from preparation to processing[J]. Progress in Polymer Science, 2003, 28(11): 1539-1641.
[2] ABEDI S, ABDOUSS M. A review of clay-supported Ziegler-Natta catalysts for production of polyolefin/clay nanocomposites through in situ polymerization[J]. Applied Catalysis A General, 2014, 475(11): 386-409.
[3] RONG J F, LI H Q, JING Z H, et al. Novel organic/inorganic nanocomposite of polyethylene. I. preparation via in situ polymerization approach[J]. Journal of Applied Polymer Science, 2001, 82(8): 1829-1837.
[4] RONG J F, JING Z H, LI H Q, et al. A polyethylene nanocomposite prepared via in-situ polymerization[J]. Macromolecular Rapid Communications, 2001, 22(5): 329-334.
[5] RONG J F, SHENG M, Li H Q, et al. Polyethylene-palygorskite nanocomposite prepared via in situ coordinated polymerization[J]. Polymer Composites 2002, 23(4): 658-665.
[6] RAMAZANI A S A, TAVAKOLZADEH M, BANIASADI H, et al. In situ polymerization of polyethylene/clay nanocomposites using a novel clay-supported Ziegler-Natta catalyst[J]. Polymer Composites 2002, 30(10): 1388-1393.
[7] CUI L Q, CHO H Y, SHIN J W, et al. Polyethylene-montmorillonite nanocomposites: Preparation, characterization and properties[J]. Macromolecular Symposia 2007, 260: 49-57.
[8] JIN Y H, PARK H J, IM S S, et al. Polyethylene/clay nanocomposite by in-situ exfoliation of montmorillonite during Ziegler-Natta polymerization of ethylene[J]. Macromolecular Rapid Communications 2002, 23(2): 135-140.
[9] YANG F, ZHANG X Q, ZHAO H C, et al. Preparation and properties of polyethylene/ montmorillonite nanocomposites by in situ polymerization[J]. Journal of Applied Polymer Science, 2003, 89(13): 3680-3684.
[10] HE F A, ZHANG L M, YANG F, et al. Polyethylene nanocomposites obtained from in-situ polymerization using supported Ziegler-Natta catalyst system[J]. Journal of Macromolecular Science Part A Pure and Applied Chemistry, 2003, 44(1): 11-15.
[11] NIKKHAH S J, RAMAZANI S A A, BANIASADI H, et al. Investigation of properties of polyethylene/clay nanocomposites prepared by new in situ Ziegler-Natta catalyst[J]. Materials & Design, 2009, 30(7): 2309-2315.
[12] CUI L Q, WOO S I, et al. Preparation and characterization of polyethylene(PE)/clay nanocomposites by in situ polymerization with vanadium-based intercalation catalyst[J]. Materials & Design, 2009, 61(4): 453-460.
[13] ABEDI S, ABDOUSS M, NEKKOMANESH-HAGHIFHI M, et al. New clay-supported ZieglerNatta catalyst for preparation of PE/clay nanocomposites via insitu polymerization[J]. Journal of Applied Polymer Science, 2013, 128(3): 1879-1884.
[14] ABEDI S, ABDOUSS M, NEKKOMANESH-HAGHIFHI M,et al. PE/clay nanocomposites produced via in situ polymerization by highly active clay-supported Ziegler-Natta catalyst[J].Polymer Bulletin, 2013, 70(4): 1313-1325.
[15] ABEDI S, ABDOUSS M, NEKKOMANESH-HAGHIFHI M, et al. Highly exfoliated PE/Na+MMT nanocomposite produced via in situ polymerization by a catalystsupported on a novel modified Na+MMT[J]. Polymer Bulletin, 2013, 70(10): 2783-2792.
[16] WANG J, LIU Z Y, GUO C Y, et al. Preparation of a PE/MT composite by copolymerization of ethylene with in-situ produced ethylene oligomers under a dual functional catalyst system intercalated into MT layer[J]. Macromolecular Rapid Communications, 2001, 22(17): 1422-1426.
[17] HUANG Y J, YANG K F, DONG J Y, et al. Copolymerization of ethylene and 10-undecen-1-ol using a montmorillonite-intercalated metallocene catalyst: Synthesis of polyethylene/montmorillonite nanocomposites with enhanced structural stability[J]. Macromolecular Rapid Communications, 2006, 27(15): 1278-1283.
[18] HUANG Y J, QIN Y W, DONG J Y, et al. PE/OMMT nanocomposites prepared by in situ polymerization approach: Effects of OMMT-intercalated catalysts and silicate modifications[J]. Journal of Applied Polymer Science, 2012, 123(5): 3106-3166.
[19] HUANG Y J, QIN Y W, DONG J Y, et al. PE/OMMT nanocomposites catalyzed by the OMMT Intercalated Et[Ind]2ZrCl2in slurry polymerization: Effects of organic solvent on ethylene polymerization behaviors and the nanocomposite structures[J]. Journal of Applied Polymer Science, 2011, 119(1): 190-200.
[20] CARRERO A, VAN GRIEKEN R, SUAREA I, et al. Development of a new synthetic method based on in situ strategies for polyethylene/clay composites[J]. Journal of Applied Polymer Science, 2012, 126(3): 987-997.
[21] WEI L M, TANG T, HUANG B T, et al. Development of a new synthetic method based on in situ strategies for polyethylene/clay composites[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2004, 42(4): 941-949.
[22] LIU C B, TANG T, WANG D, et al. In situ ethylene homopolymerization and copolymerization catalyzed by zirconocene catalysts entrapped inside functionalized montmorillonite[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2003, 41(14): 2187-2192.
[23] LIU C B, TANG T, HUANG B T, et al. Zirconocene catalyst well spaced inside modified montmorillonite for ethylene polymerization: Role of pretreatment and modification of montmorillonite in tailoring polymer properties[J]. Journal of Catalysis, 2004, 221(1): 162-169.
[24] LEONE G, BERTINI F, CANETTI M, et al. In situ polymerization of ethylene using metallocene catalysts: Effect of clay pretreatment on the properties of highly filled polyethylene nanocomposites[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2008, 46(16): 5390-5403.
[25] XALTER R, HALBACH TS, MULHAUPT M, et al. New polyolefin nanocomposites and catalyst supports based on organophilic boehmites[J]. Macromolecular Symposia, 2006, 236: 145-150.
[26] MANESHI A, SOARES J B P, SIMON L C, et al. Polyethylene/clay nanocomposites made with metallocenes supported on different organoclays[J]. Macromolecular Chemistry and Physics, 2011, 212: 216-228.
[27] HEINWMANN J, REICHERT P, THOMANN R, et al. Polyolefin nanocomposites formed by melt compounding and transition metal catalyzed ethene homo- and copolymerization in the presence of layered silicates[J]. Macromolecular Rapid Communications, 1999, 20(8): 423-430.
[28] HE F A, ZHANG L M, JIANG H L, et al. A new strategy to prepare polyethylene nanocomposites by using a late-transition-metal catalyst supported on AlEt3-activated organoclay[J]. Macromolecular Rapid Communications, 2007, 67(7-8): 1727-1733.
[29] HE F A, ZHANG L M. Using inorganic POSS-modified laponite clay to support a nickel alpha-diimine catalyst for in situ formation of high performance polyethylene nanocomposites[J]. Nanotechnology, 2007, 17(24): 1727-1733.
[30] HE F A, ZHANG L M. Study on branching structure, melting, and crystallization of polyethylene prepared by nickel a-diimine catalyst covalently intercalated inside OapPOSS-modified laponite clay gallery[J]. Polymer Testing, 2014, 35(24): 80-86.
[31] SHIN S Y A, SIMON L C, SOARES J B P, et al. Polyethylene-clay hybrid nanocomposites: In situ polymerization using bifunctional organic modifiers[J]. Polymer, 2003, 44(18): 5317-5321.
[32] RAY S, GALGALI G, SIVARAM S, et al. In situ polymerization of ethylene with bis(imino)pyridine iron(II) catalysts supported on clay: The synthesis and characterization of polyethylene-clay nanocomposites[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2005, 43(2): 304-318.
[33] MIGNONI M L, MAERINS S J V, DE SOUZA M O, et al. Polyethylene-montmorillonite nanocomposites obtained by in situ polymerization of ethylene with nickel-diimine catalysts[J]. Journal of Applied Polymer Science, 2011, 122(3): 2159-2165.
[34] SCOTT S L, PEOPLES B C, YUNG C, et al. Highly dispersed clay-polyolefin nanocomposites free of compatibilizers, via the in situ polymerization ofα-olefins by clay-supported catalysts[J]. Chemical Communicatoins, 2008, (35): 4186-4188.
[35] LEONE G, BERTINI F, CANETTI M, et al. Long-lived layered silicates-immobilized 2,6-bis(imino)pyridyl iron (II) catalysts for hybrid polyethylene nanocomposites by in situ polymerization: effect of aryl ligand and silicate modification[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2009, 47(2): 548-564.
[36] TONG X, LIU C, CHENG H M, et al. Surface modification of single-walled carbon nanotubes with polyethylene via in situ Ziegler-Natta polymerization[J]. Journal of Applied Polymer Science, 2004, 92(6): 3697-3700.
[37] AMOLI B M, RAMAZANI S A, AHMAD L H, et al. Surface modification of single-walled carbon nanotubes with polyethylene via in situ Ziegler-Natta polymerization[J]. Journal of Applied Polymer Science[J]. 2012, 125(S1): E453-E456.
[38] JAFAR S, RAMAZANI S A, FARHAD K, et al. Thermal degradation behavior and kinetic analysis of ultra high molecular weight polyethylene based multi-walled carbon nanotube nanocomposites prepared via in-situ polymerization[J]. Journal of Macromolecular Science Part A Pure and Applied Chemistry, 2012, 49(9): 749-757.
[39] PARK H J, KIM J H, SEO Y S, et al. Wear behavior of in situ polymerized carbon nanotube/ultra high molecular weight polyethylene composites[J]. Macromolecular Research, 2013, 21(9): 965-970.
[40] LIU Z, YU M S, WANG J, et al. Preparation and characterization of novel polyethylene/carbon nanotubes nanocomposites with core-shell structure[J]. Journal of Industrial and Engineering Chemistry, 2014, 20(4): 1804-1811.
[41] BONDUEL D, MAIANIL M L, ALEXANDRE M, et al. Supported coordination polymerization: A unique way to potent polyolefin carbon nanotube nanocomposites[J]. Chemical Communications, 2005, (6): 781-783.
[42] BONDUEL D, BREDEAU S, ALEXANDRE M, et al. Supported metallocene catalysis as an efficient tool for the preparation of polyethylene/caron nanotube nanocomposites:Effect of the catalytic system on the coating morphology[J]. Journal of Materials Chemistry, 2007, 17(22): 2359-2366.
[43] LI S Y, CHEN H, CUI D M, et al. Structure and properties of multi-walled carbon nanotubes/polyethylene nanocomposites synthesized by in situ polymerization with supported Cp2ZrCl2catalyst[J]. Polymer Composites, 2010, 31(3): 507-515.
[44] TANG J J, LI S Y, WANG Y H, et al. In situ ethylene copolymerization with an olefin-type monomer for one-pot synthesis of polyethylene tethered on multi-walled carbon nanotubes[J]. Journal of Industrial and Engineering Chemistry, 2013, 31(10): 1329-1333.
[45] KIM J H, SEO Y S, HONG S M, et al. Preparation of PE/MWNT nanocomposites by in-situ metallocene polymerization[J]. International Journal of Material Forming[J], 2009, 2(1): 873-875.
[46] KIM J H, HONG S M, KWAK S J, et al. Physical properties of nanocomposites prepared by in situ polymerization of high-density polyethylene on multiwalled carbon nanotubes[J]. Physical Chemistry Chemical Physics, 2013, 11(46): 10851-10859.
[47] KIM J H, KWAK S J, HONG S M, et al. Nonisothermal crystallization behaviors of nanocomposites prepared by in situ polymerization of high-density polyethylene on multiwalled carbon nanotubes[J]. Macromolecules, 2010, 43(24): 10545-10533.
[48] BAHULEYAN B K, ATIEH M A, DE S K, et al. Easy one-pot method to control the morphology of polyethylene/carbon nanotube nanocomposites using metallocene catalysts[J]. Journal of Polymer Research, 2012, 19(2): 9744.
[49] DONG X C, WANG L, SUN T X, et al. Study on ethylene polymerization catalyzed by Cp2ZrCl2/carbon nanotube system[J]. Journal of Molecular Catalysis A-Chemical, 2006, 255(1-2): 10-15.
[50] DONG X C, WANG L, DONG L B, et al. Preparation of nano-polyethylene fibres using Cp2ZrCl2/carbon nanotube catalytic system[J]. Materials Letters, 2007, 61(14-15): 3111-3115.
[51] TRUJILLO M, AENAL M L,MULLER A J, et al. Thermal and morphological characterization of nanocomposites prepared by in-situ polymerization of high-density polyethylene on carbon nanotubes[J]. Macromolecules, 2007, 40(17): 6268-6276.
[52] TOTI A, GIAMBASTIANI G,BIANCHINI C, et al. Tandem action of early-late transition metal catalysts for the surface coating of multiwalled carbon nanotubes with linear low-density polyethylene[J]. Chemistry of Materials, 2007, 20(9): 3092-3098.
[53] PARK S J, YOON S W,CHOI H C, et al. Pristine multiwalled carbon nanotube/polyethylene nanocomposites by immobilized catalysts[J]. Chemistry of Materials, 2008, 20(14): 4588-4594.
[54] PARK S J, CHOI I S. Production of ultrahigh-molecular-weight polyethylene/pristine MWCNT composites by half-titanocene catalysts[J]. Advanced Materials, 2009, 21(8): 902-905.
[55] ZHANG L P, ZHANG W J, SERP P, et al. Ethylene polymerization catalyzed by pyrene-tagged iron complexes: the positive effect ofπ-conjugation and immobilization on multiwalled carbon nanotubes[J]. Chem Cat Chem, 2014, 6(8): 1310-1316.
[56] 何富安. 新型聚乙烯纳米复合材料的制备与性能研究[D].广州:中山大学化学与化学工程学院. 2007.
[57] VEGA J F, MARTINEZ-SALAZAR J, TRUJILLO M, et al. Rheology, processing, tensile properties, and crystallization of polyethylene/carbon nanotube nanomposites[J]. Macromolecules, 2009, 42(13): 4719-4272.
[58] VEGA J F, SILVA D Y, VIXWNRW-AKUQUE E, et al. Influence of chain branching and molecular weight on melt rheology and crystallization of polyethylene/carbon nanotube nanocomposites[J]. Macromolecules, 2014, 47(16): 5668-5681.
[59] BREDEAU S, BOGGIONI L, BERTINI F, et al. Ethylene-norbornene copolymerization by carbon nanotube-supported metallocene catalysis: generation of high-performance polyolefinic nanocomposites[J]. Macromolecular Rapid Communications, 2007, 28(7): 822-827.
[60] RAVASIO A, BOGGIONI L, TRITTO I, et al. A non-PFT (polymerization filling technique) approach to poly(ethylene-co-norbornene)/MWNTs nanocomposites by in situ copolymerization with scandium half-sandwich catalyst[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2009, 47(21): 5709-5719.
[61] ALEXANDRE M, PLUTA M, DUBOIS P, et al. Metallocene catalyzed polymerization of ethylene in the presence of graphite, 1-Synthesis and characterization of the composites[J]. Macromolecular Chemistry and Physics, 2001, 202(11): 2239-2246.
[62] PLUTA M, ALEXANDRE M, BLACHER S, et al. Metallocene-catalyzed polymerization of ethylene in the presence of graphite II Structure and electrical properties of the composites[J]. Polymer, 2001, 42(22): 9293-9300.
[63] FABIANA D C F, JONATHAN M G, NaRA S B, et al. Polyethylene/graphite nanocomposites obtained by in situ polymerization[J]. Journal of Polymer Science Part A-Polymer Chemistry, 2010, 48(3): 692-698.
[64] FABIANA D C F, NARA S B, ANA P G, et al. Thermal, electrical, and mechanical properties of polyethylene-graphene nanocomposites obtained by in situ polymerization[J]. Journal of Applied Polymer Science . 2012, 128(5): 2630-2637.
[65] HU Z, LIU C B. Polyethylene/graphite oxide nanocomposites obtained by in situ polymerization using modified graphite oxide-supported metallocene catalysts[J]. Journal of Polymer Research. 2013, 20(1): 39.
[66] LEE J S, KO Y S. Synthesis of petaloid graphene/polyethylene composite nanosheet produced by ethylene polymerization with metallocene catalyst adsorbed on multilayer graphene[J]. Catalysis Today. 2014, 232: 82-88.
[67] TODD A D, BIELAWSKI C W. Thermally reduced graphite oxide reinforced polyethylene composites: A mild synthetic approach[J]. Polymer, 2013, 54(17): 4427-4430.
[68] STURZEL M, KEMPE F, THOMMAN Y, et al. Novel graphene UHMWPE nanocomposites prepared by polymerization filling using single-site catalysts supported on functionalized graphene nanosheet dispersions[J]. Macromolecules, 2012, 45 (17): 6878-6887.
[69] CHOI B, LEE J, LEE S J, et al. Generation of ultra-high-molecular-weight polyethylene from metallocenes immobilized onto N-Doped graphene nanoplatelets[J]. Macromolecules, 2013, 34 (6): 533-538.
[70] ALEXANDRE M, MARTIN E, DUBOIS P, et al. Use of metallocenes in the polymerization-filling technique with production of polyolefin-based composites[J]. Macromolecular Rapid Communications, 2000, 21(13): 931-936.
[71] ALEXANDRE M, MARTIN E, DUBOIS P, et al. Polymerization-filling technique: An efficient way to improve the mechanical properties of polyethylene composites[J]. Chemistry of Materials, 2001, 13(2): 236-237.
[72] HE F A, ZHANG L M. New polyethylene nanocomposites prepared by in-situ polymerization method using nickel a-diimine catalyst supported on organo-modified ZnAl layered double hydroxide[J]. Composites Science and Technology, 2007, 67(15-16): 3226-3232.
[73] HE F A, ZHANG L M. Organo-modified ZnAl layered double hydroxide as new catalyst support for the ethylene polymerization[J]. Journal of Colloid and Interface Science, 2007, 315(2): 439-444.
[74] MOBTEIL V, STUMBAUM J, THOMANN R, et al. Silica/polyethylene nanocomposite particles from catalytic emulsion polymerization[J]. Macromolecules. 2006, 39 (6): 2056-2062.
[75] WANG X, WU Q Y, DONG J Y, et al. Characterization of polyethylene/kaolin composites by polymerization filling with Cp2ZrCl2/MAO catalyst system[J]. Journal of Applied Polymer Science, 2002, 85(14): 2913-2921.
[76] ZHANG F, KARAKI T, ADACHI M, et al. Preparation of ferroelectric (Pb,Sr)TiO3/polyethylene nanocomposites and their dielectric properties[J]. Japanese Journal of Applied Physics Part 1-Regular Papers Brief Communications & Review Papers. 2006, 45 (3A): 1873-1876.
[77] JONGSOMJIT B, PANPRANOT J, PRASERTHDAM P. Effect of nanoscale SiO2and ZrO2as the fillers on the microstructure of LLDPE nanocomposites synthesized via in situ polymerization with zirconocene[J]. Materials Letters. 2007, 61 (6): 1376-1379.
[78] CHAICHANA E, JONGSOMJIT B, PRASERTHDAM P. Effect of nano-SiO2particle size on the formation of LLDPE/SiO2nanocomposite synthesized via the in situ polymerization with metallocene catalyst[J]. Chemical Engineering Science, 2007, 62 (3): 899-905.
[79] JONGSOMJIT B, PANPRANOT J, PRASERTHDAM P. LLDPE/nano-silica composites synthesized via in situ polymerization of ethylene/1-hexene with MAO/metallocene catalyst[J]. Journal of Materials Science, 2005, 40(8): 2043-2045.
[80] JONGSOMJIT B, PANPRANOT J, OKADA M, et al. Characteristics of LLDPE/ZrO2nanocomposite synthesized by in-situ polymerization using a zirconocene/MAO catalyst[J]. Iranian Polymer Journal, 2006, 15(5): 433-439.
[81] DESHARUN C, JONGSOMJIT B, PPRASERTHDAM P, et al. Study of LLDPE/alumina nanocomposites synthesized by in situ polymerization with zirconocene/d-MMAO catalyst[J]. Catalysis Communications. 2008, 9(4): 522-528.
[82] CHAICHANA E, NGOWTHANAWAT P, MEKASUWANDUMRONG O, et al. Catalytic performance of ZnO nanoparticle in formation of LLDPE/ZnO nanocomposites[J]. Iranian Polymer Journal, 2012, 21(1): 51-63.
[83] CHAICHANA E, PATHOMSAP S, MEKASUWANDUMRONG O, et al. LLDPE/TiO2nanocomposites produced from different crystallite sizes of TiO2via in situ polymerization[J]. Chinese Science Bulletin, 2012, 57(17): 2177-2184.
[84] CHENG W X, MIAO W, PENG J, et al. Synthesis of silica/polyolefin nanocomposites via two-step method[J]. Iranian Polymer Journal, 2009, 18(5): 365-371.
[85] COVARRUBIASA C, QUIJADAA R. Preparation of aluminophosphate/polyethylene nanocomposite membranes and their gas permeation properties[J]. Journal of Membrane Science. 2010, 358(1-2): 33-42.
[86] WANG N, SHI Z X, PENG J, et al. The influence of modification of mesoporous silica with polyethylene via in situ Ziegler-Natta polymerization on PE/MCM-41 nanocomposite[J]. Journal of Composite Materials, 2008, 42(12): 1151-1157.
[87] KALEEL S H A, BAHULEYAN B K, MASIHULLAH J, et al. Thermal and mechanical properties of polyethylene/doped-TiO2nanocomposites synthesized using in situ polymerization[J]. Journal of Nanomaterials, 2011: 964353.
[88] WANG L, FENG L X, XIE T. Novel magnetic polyethylene nanocomposites produced by supported nanometre magnetic Ziegler-Natta catalyst[J]. Polymer International, 2000, 49(2): 184-188
[89] WANG L, YUAN Y L, FENG L X. Preparation of novel magnetic polyethylene through polymerization in situ using CrxFe3-xO4/AlR3/TiCl4supported nanometer magnetic Ziegler-Natta catalyst[J]. Polymer Journal, 1999, 49(12): 1281-1283.
[90] WANG L, FENG L X, YANF S L. Studies on the preparation of new magnetic polyolefins using nanometer magnetic Ziegler-Natta catalyst[J]. Journal of Applied Polymer Science. 1999, 71(12): 2807-2090.
[91] 江洪流, 魏珊珊, 胡扬剑, 等. 负载型磁性纳米二亚胺镍催化剂制备新型磁性支化聚乙烯[J]. 高分子材料科学与工程. 2007, 23(2): 246-249.(JIANG Hongliu, WEI Shanshan, HU Yangjian, et al. Preparation of branched magnetic polyethylenes using nickelα-diimine complex covalently supported on nanometer magnetic support[J].Polymer Materials Science and Engeering, 2007,23(2):246-249.)
[92] PARK H J, KWAK S Y, KWAK S, et al. Wear-resistant ultra high molecular weight polyethylene/zirconia composites prepared by in situ Ziegler-Natta polymerization[J]. Macromolecular Chemistry and Physics. 2005, 18(9): 945-950.
[93] POTSCHKE P, BHATTAXHARYYA A R, JANKE A. Carbon nanotube-filled polycarbonate composites produced by melt mixing and their use blends with polyethylene[J]. Carbon, 2004,42(5-6):965-969.
A Review for Preparation of High Performance Polyethylene-Based Composites by In-situ Polymerization
HE Fuan1, 2, ZHANG Liming1
(1.InstituteofPolymerScience,CollegeofChemistryandChemicalEngineering,SunYat-senUniversity,Guangzhou510275,China;2.CollegeofChemicalEngineering,GuangdongUniversityofPetrochemicalTechnology,Maoming525000,China.)
In this review, recent progress on the preparation of high performance polyethylene-based composites by in-situ polymerization was commented, involving mainly clay/polyethylene composites, carbon nanotubes/polyethylene composites and graphite-based fillers/polyethylene composites. In particular, some routes to the in-situ polymerization in the presence of supported catalysts were summarized. The morphologies, structure and properties of resultant polyethylene-based composites were also discussed. In addition, some challenges for the development of high performance polyethylene-based composites were pointed out.
polyethylene; composites; in-situ polymerization; modifying agents
2014-10-20
广东省自然科学基金研究团队项目(039184)和教育部新世纪优秀人才支持计划项目(NCET-04-0810)资助 第一作者: 何富安,男,研究员,博士,从事高分子材料设计制备与性能研究
张黎明,男,教授,博士,从事高分子材料设计制备与性能研究;Tel:020-84112354;E-mail:ceszhlm@mail.sysu.edu.cn
1001-8719(2015)02-0369-21
O63
A
10.3969/j.issn.1001-8719.2015.02.017
