多相Mo基催化剂上烯烃歧化反应
2015-06-24李秀杰张大洲辛文杰朱向学陈福存谢素娟刘盛林徐龙伢
李秀杰,张大洲,2,辛文杰,朱向学,陈福存,谢素娟,刘盛林,徐龙伢
(1.中国科学院 大连化学物理研究所, 辽宁 大连 116023;2.中国五环工程有限公司, 湖北 武汉 430223)
多相Mo基催化剂上烯烃歧化反应
李秀杰1,张大洲1,2,辛文杰1,朱向学1,陈福存1,谢素娟1,刘盛林1,徐龙伢1
(1.中国科学院 大连化学物理研究所, 辽宁 大连 116023;2.中国五环工程有限公司, 湖北 武汉 430223)
烯烃歧化是一种通过C—C键断裂重排生成新的烯烃分子的反应,可以根据市场需求灵活调控目标产物的生成,是烃类资源高效转化利用的一条重要途径。与均相催化剂相比,烯烃歧化多相催化体系具有易制备、分离简单、可循环再生的优点,在大规模化工生产中具有很好的应用前景。从催化剂的制备方法、应用反应类型及反应机理方面,重点阐述了多相Mo基催化剂催化烯烃歧化反应的研究现状和最新进展。
烯烃歧化;多相催化;Mo;复合载体
烯烃歧化反应,又称烯烃易位反应、烯烃复分解反应,是由不饱和的C=C或C≡C在催化剂的作用下发生C骨架重排的反应。该反应能使通常呈化学惰性的双键或三键彼此偶联,极大地丰富了人们在合成新的C—C骨架时的想象空间。自从Banks和Bailey于1964年发现该反应以来,在均相催化体系和多相催化体系均得到了迅速发展和应用[1-4]。目前,该反应已经广泛应用于石油化工、有机化学、高分子化学领域,并成为合成化工基本原料、精细化学品、天然产物、新型药物、生物活性化合物等产品的一条重要技术路线。
多相烯烃歧化反应催化剂通常由过渡金属氧化物或者有机过渡金属化合物分散在高比表面积的无机氧化物载体上制得。根据过渡金属的类型主要分为Re基、W基和Mo基三大类。Re基催化剂在低温下具有很高的歧化活性,缺点在于金属Re价格昂贵,且Re2O7在催化剂制备过程中易升华,这在一定程度上限制了其广泛应用性[5]。W基催化剂在歧化反应中通常需要较高的反应温度,歧化活性相对较低,但是对水、空气等杂质不敏感,是目前唯一成功应用于工业装置的催化剂[6]。Mo基催化剂歧化活性介于Re基、W基之间,反应温度适中。笔者重点阐述多相Mo基催化剂催化烯烃歧化反应机理,总结了催化剂制备方法、载体性质影响以及多相歧化反应的类型。
1 多相Mo基歧化催化剂的制备方法
1.1 传统浸渍法
负载型歧化催化剂的制备通常采用浸渍法(Wet impregnation)。将具有一定表面性质的载体放入含有活性组分的溶液中浸泡,借助毛细管压力使活性组分渗透到载体空隙内部[7-10]。采用传统浸渍法制备负载型催化剂时,可以使用现成的具有一定外形和尺寸的载体材料,所浸渍的组分几乎能全部负载到载体表面,因此,可以有效控制金属中心的负载量。采用该方法,金属组分利用率高,对以贵金属作为活性组分的材料尤为重要。但是由于活性组分离子与载体间复杂的相互作用,可能使得金属离子不能均匀扩散至孔道内部,致使金属物种在载体上存在的状态不均一。此外,若载体与活性组分溶液间存在较强的相互作用,还可能形成对歧化反应不利的物种,如钼酸铝等[11-12]。
1.2 热分散法
热分散法(Thermal spread method)是将金属组分(通常为氧化物形式)与载体直接经过高温焙烧处理的方法。与浸渍法相比,该方法的优点是避免使用溶剂,减去干燥过程,具有较好的经济和环境效益。
Debecker等[13-14]对比分析了浸渍法和热分散法制备的Mo负载型歧化催化剂。结果表明,对于浸渍法制备的Mo催化剂,在固-液界面处有[AlMo6]物种生成,该物种经过干燥、焙烧后形成无活性的晶相MoO3和钼酸铝,限制了催化剂的歧化活性;对于采用直接热分散方法制备的Mo催化剂,MoO3晶相会自分散到载体表面和孔内,并形成无定型的表面层Mo物种(歧化活性位),尤其是在低Mo负载量时Mo物种可以有效分散于载体表面,而且没有晶相MoO3和钼酸铝物种的形成;然而当Mo负载量较高时,部分MoO3未能充分分散,形成了少量MoO3微晶,抑制了催化剂的歧化活性。
对于介孔MCM-41等载体,若采用浸渍法,钼酸铵溶液容易破坏其六方介孔结构。为此,Balcar等[15-17]在制备介孔硅基载体负载的歧化催化剂时多采用热分散法。作者曾以MoO2(acac)2和Mo2(gly)2为Mo源,采用热分散的方法将Mo物种负载到介孔SBA-15、MCM-41等材料上制得Mo基催化剂,将其催化1-辛烯歧化反应,取得较好的效果[16]。
1.3 溶胶-凝胶一步合成法
采用溶胶-凝胶法制备负载型歧化催化剂,可以提高金属与载体间的作用力,改善活性物种的分散度,同时得到的催化剂具有大表面积、特定孔道结构等特性。Hu等[18]和赵秦峰等[19]以P123为模板剂,分别以正硅酸乙酯(TEOS)和钨酸钠(NaWO4·2H2O)为Si源和W源,在酸性条件下采用一步溶胶-凝胶方法制备了W取代的介孔SBA-15催化剂。光谱分析表明,W物种高度分散于介孔分子筛Si骨架内。采用该方法制备的n(Si)/n(W)=30的W催化剂催化1-丁烯自歧化反应(350℃,0.5 MPa),丙烯收率是传统浸渍法制备的WO3/SiO2催化剂的2倍以上;当催化剂的n(Si)/n(W)低于30时,表面W物种的聚集和介孔结构的部分坍塌,导致催化剂催化1-丁烯自歧化反应转化率和丙烯选择性均有所下降[18]。
传统的溶剂-凝胶法是基于醇盐的水解和缩聚。然而,对于SiO2-Al2O3-MoO3三元混合物而言,不同组分前驱体的反应速率往往不同,因此很难同时控制凝胶的组成和结构。即使对于简单的二元混合物,制备均一的介孔材料也需要复杂的实验流程,例如活性差的前驱体的预水解处理,或用特殊螯合剂钝化活性较强的组分等[11,20]。近来,Debecker等[21-22]以SiCl4、AlCl3和MoCl5为原料,以二异丙醚为溶剂,采用直接非水溶性溶胶-凝胶方法制备了具有介孔结构的SiO2-Al2O3-MoO3三元混合氧化物(如图1(a)所示)。结合相关表征发现,采用该方法制备的催化剂样品中Mo物种能得到更好的分散,其配位环境与传统浸渍法制备的Mo基催化剂明显不同。采用溶胶-凝胶法或传统浸渍法制备的样品中均检测到孤立的Mo离子,但浸渍法制备的样品中同时也检测到多聚态Mo离子。将这2种方法制备的催化剂用于丙烯歧化反应时发现,在低Mo负载量下,浸渍法制备的催化剂表面存在更多的活性Mo物种,使得其活性较高;高Mo负载量下,浸渍法制备的催化剂上出现晶相MoO3,导致活性不再升高,非水溶性溶胶-凝胶法制备的样品上Mo物种呈高度分散状态,从而使得该催化剂活性继续提高[21]。
最近,Debecker等[23]又将含有模板剂的溶胶-凝胶法与气溶胶处理技术结合,通过气溶胶的快速干燥控制溶胶-凝胶过程的动力学,最终得到具有高比表面积、孔道结构有序、Mo物种高度分散且具有较高歧化活性的SiO2-Al2O3-MoO3三元催化剂。采用相同方法,只将活性金属组分换为W物种后制备的催化剂同样具有较高的乙烯和2-丁烯交叉歧化活性[24]。

图1 非水溶性溶胶-凝胶法制备步骤及歧化活性评价[21]
1.4 高温喷雾热分解法
高温喷雾热分解法(FSP)是在高温炉内将原料溶液,如金属盐溶液雾化,然后使其瞬间发生高温热分解(包括反应、高温焙烧),从而得到粉体产品的过程。Debecker等[25]采用FSP法制备了Al2O3-SiO2负载的Mo基歧化催化剂。在制备过程中,将特定性质的Al源、Si源、Mo源与二甲苯混合均匀配成溶液,随后通入FSP反应器生成催化剂粉末。通过该方法可以制备具有特殊结构和聚集状态的负载型催化剂,便于研究反应机理,但受原料和合成设备的限制,实际生产成本太高。
1.5 均相催化剂多相化
传统的多相催化剂活性位点不均一,种类多,不利于研究某一活性位与反应性能的关系,限制催化剂的进一步设计改善;而均相催化体系结构明确,可以有效确定活性位与反应性能间的关系,从分子水平上改进催化剂。但均相催化剂存在回收难、产物分离成本高等缺点。基于此,研究者们开始利用表面金属有机化学的方法实现均相与多相歧化催化体系的优势互补,同时在分子水平上理解多相催化剂的歧化反应机理。该工作是目前学术界的研究热点之一。
法国的J.M.Basset教授和瑞士ETH Zürich的C.Coperet教授在均相歧化催化剂的多相化方面进行了深入而系统的研究[26-28]。根据结构-性能关系,从分子上设计催化剂可以分成三步,首先采用各种表征方法,在分子水平上确定表面物种的结构;其次,选择和控制表面化学反应,产生特定的表面活性位和实现催化活性需要的关键配体,如在载体的制备过程中控制表面羟基的浓度(焙烧、脱羟基、表面改性等),通过将其与结构确定的配合物反应生成特定的表面活性位;最后,评价催化剂的反应性能,根据结构-性能的关系得出假设,随后通过失活机理和表面修饰指导催化剂的改善,如图2所示[27]。
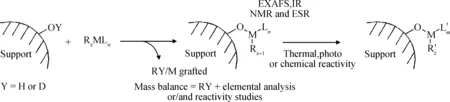
图2 均相催化剂多相化的构效关系示意图[27]
2 载体性质对Mo基催化剂催化烯烃歧化反应的影响
负载型歧化催化剂由活性金属组分和载体两部分组成。载体的性质(包括酸性、比表面积、孔道结构、表面官能团分布等)会影响Mo物种的落位及与载体的相互作用,进而对催化剂的反应活性产生很大影响。
2.1 载体酸性
大多数研究认为,载体酸性对烯烃歧化反应有利。但酸性位分为Brönsted和Lewis酸位,酸类型不同对反应的影响机制也不同,因此具体的研究结论具有很大的不确定性。

载体的Brönsted酸位和Lewis酸位对金属中心结构和歧化反应存在不同的影响。Li等[32-35]在考察分子筛-Al2O3复合载体负载的Mo基催化剂催化烯烃歧化反应性能时发现,Mo物种优先与Hβ-Al2O3复合载体上的Lewis酸位相互作用,剩余Brönsted酸性位较多;Mo物种与Hβ分子筛之间存在强烈的相互作用,Mo物种的引入导致分子筛骨架破坏,骨架Al被抽出形成钼酸铝物种,酸位数目大大降低,造成Mo/Hβ催化剂活性很低。反应评价结果表明,只有适当的Brönsted酸性位才有利于歧化反应,同时抑制异构化、聚合等副反应的发生[32-33]。此外,通过对催化剂进行水蒸气后处理以及添加Mg助剂改性后发现,移除催化剂上的弱酸位并不会影响歧化反应初始活性,反而可以抑制齐聚反应的发生,提高催化剂的稳定性,但是Brönsted酸性位的减少会直接导致催化剂活性降低[36]。
金属中心与Lewis酸位作用后有可能会生成歧化活性中心。例如,Vanroosmalen等[37-38]通过用六甲基二硅氮烷硅烷化WO3/SiO2催化剂表面的弱Brönsted酸性羟基,同时借助W周围形成吸电子≡Si—O基团可以提高W周围的Lewis酸性(易吸附富含电子的烯烃),从而提高了催化剂的歧化活性。Moses等[39-40]将CH3ReO3负载到SiO2-Al2O3载体表面上制得Re基催化剂,实验发现,高Re负载量下催化剂的活性与低Re负载量下催化剂的活性类似(将CH3ReO3负载到SiO2上则完全没有歧化活性),但高Re负载量的催化剂所得产物的选择性较高,低Re负载量的催化剂会发生由SiO2-Al2O3载体上的酸性羟基催化的烯烃异构化、齐聚副反应,使得产物的选择性较低。结合表征结果,作者认为,低Re负载量下Re会优先与载体上的Lewis酸位作用,生成有利于歧化反应的活性物种;高Re负载量下,Re会通过氢键与SiO2-Al2O3载体上的表面羟基相互作用,生成无活性的物种,同时会减少表面的酸性位,在一定程度上抑制了异构化、齐聚副反应的发生。但作者用六甲基二硅氮烷硅烷化修饰SiO2-Al2O3载体后发现,低Re负载量下的催化剂也具有高Re负载量时所具有的活性,同时载体经修饰后表面的Brönsted酸位较少,副反应少,产物选择性提高[39]。
也有研究者认为,Lewis酸位对歧化反应的引发具有重要作用。例如Schekler-Nahama等[41]采用Py-IR实验表征Re2O7/Al2O3催化剂上的Lewis酸位和Brönsted酸位时发现,提高Re负载量和焙烧温度均能提高催化剂上Lewis酸位数量,且表现出了较高的丙烯歧化活性。结合原位FT-IR和氨毒化实验结果,吸附的氨气经过150℃脱附后,催化剂表面仅有Brönsted酸位,Lewis酸位被氨覆盖,原位红外光谱没有检测到产物乙烯的红外吸收峰;而当脱附温度升至250℃时,催化剂上的Lewis酸位恢复,同时检测到产物乙烯的红外吸收峰。
此外,理论计算也表明载体酸性对催化剂的歧化反应活性存在显著影响。例如,Li等[42]采用不同Si—H键长度的团簇模型来反映Hβ分子筛载体的Brönsted酸强度,通过DFT计算发现,载体酸性越强,由Mo前驱体形成Mo卡宾所需要的能量越低,初始卡宾越容易形成;虽然载体酸性对Mo卡宾催化的反应循环过程影响相对较小,但酸性提高同样会有利于反应的进行。Handzlik等[43]考察了Mo/HZSM-5体系中2种不同结构Mo物种的歧化活性。与取代1个骨架桥联羟基相比,当Mo物种取代2个桥联羟基落位后,形成的MoO(CH2)结构具有较高的正电性,说明此配位结构的载体吸电子能力较强;受载体吸电子作用的影响,2种结构Mo物种中心的电子密度降低,有利于乙烯与Mo=CH2发生环加成反应。然而,对于取代1个桥联羟基的Mo物种,其形成的钼杂环丁烷比较稳定,不利于开环形成活性卡宾中心,因此取代2个桥联羟基的Mo物种具有更好的歧化反应活性。这进一步从理论上证实了载体酸性能显著影响催化剂的歧化反应活性。
2.2 孔道结构
近年来,负载型烯烃歧化催化剂的研究新进展之一就是以介孔分子筛或介孔Al2O3为载体的介孔催化剂的制备及应用[10,16,44-46]。介孔材料一般具有规整均一的孔道结构和大比表面积,使得负载后的催化剂通常表现出比传统SiO2和Al2O3为载体的催化剂更好的活性、选择性以及更长的反应稳定性,已成为烯烃歧化催化剂的一个研究热点。目前,应用于烯烃歧化反应的介孔催化剂主要包括介孔分子筛(MCM-41、MCM-48、HMS、SBA-15等)负载的W基和Mo基催化剂,以及介孔Al2O3负载的Mo基和Re基催化剂,还包括金属有机配合物固载于介孔载体上形成的多相催化剂。
日本东京大学的Ookoshi等[47]首先报道了以六角中空介孔HMS分子筛为载体负载的Mo基歧化催化体系,其1-辛烯歧化反应活性显著高于以往的Mo/Al2O3和Mo/SiO2催化剂。此外,对比不同孔道尺寸和粒径的载体时发现,受分子传质的影响,在孔道尺寸和粒径均较小的催化剂催化1-辛烯歧化反应初始活性较低,但粒径较小的催化剂同时具有较短的孔道结构,会缩短聚合中间物种的扩散路径,防止孔道堵塞,因此该催化剂随反应的进行反而表现出较好的稳定性[47]。在随后的工作中,其他学者同样发现,介孔MCM-41、SBA-15等载体负载的Mo基或W基催化剂在烯烃歧化反应中表现出优良的反应效果[16,18,48]。
对于介孔Al2O3为载体的催化剂,以Re基催化体系的研究居多[10,46,49-52]。可以采用传统浸渍法或热分散法将NH4ReO4、HReO4、Re2O7、CH3ReO3等Re源负载于介孔Al2O3上制备得到Re基催化剂。Onaka等[52]首先发现,平均孔径为3 nm的介孔Al2O3负载的Re基催化剂催化1-辛烯和7-十四碳烯的歧化反应速率均高于传统的Re/Al2O3催化剂。作者认为,虽然2种载体上Re物种的结构相似,但是介孔Al2O3具有规整的孔道结构,有利于Re物种与孔道凹表面上的羟基和配位不饱和的Al原子同时结合,稳定活泼的金属卡宾中间物种[10]。Balcar等[15,45]研究发现,1-癸烯歧化反应活性受介孔Al2O3孔径的影响较小,但在含有极性官能团的对烯丙基苯甲醚自歧化和二烯丙基丙二酸二乙酯闭环歧化中,孔径越大,反应转化率越高,催化剂的失活速率越慢。总之,基于反应分子的尺寸和极性,通过调节介孔Al2O3的孔径在一定程度上可以改变产物的组成。
3 Mo基催化剂催化烯烃歧化反应的类型
3.1 低碳烯烃歧化
烯烃歧化工艺是由Phillips石油公司开发的丙烯歧化转化为乙烯和2-丁烯的三烯法工艺(Triolefin process)。后来,随着丙烯需求量超过乙烯,ABB Lummus公司在该工艺的基础上开发了乙烯和2-丁烯交叉歧化生产丙烯的OCT工艺(Olefins conversion technology),现已在全球得到了广泛的工业化应用,其中包括中国上海赛科石化公司建成的一套与乙烯裂解装置配套的14万t/a装置。
目前,低碳烯烃工艺的开发主要包括乙烯与2-丁烯交叉歧化制丙烯[23,32]、1-丁烯与2-丁烯歧化制丙烯[9,53]、1-丁烯自歧化制1-己烯[54]、2-丁烯(1-丁烯)与异丁烯歧化制丙烯和异戊烯[55-57]、异丁烯自歧化制四甲基乙烯[58]等。当然,为了方便实验研究和理论计算歧化反应机理,许多研究者将丙烯自歧化作为探针反应[21,59]。
针对低碳烃中的乙烯、丁烯制丙烯反应,徐龙伢等开发了Mo/Hβ-Al2O3体系[32,34,60]。与传统的Mo/Al2O3体系相比,分子筛的引入有效提高了催化剂的低温歧化活性[32,34,61-62]。所开发的Mo/Hβ-Al2O3催化剂在100 mL催化剂规模装置上实现了在30~60℃低温反应条件下,丁烯转化率高于70%,丙烯选择性大于90%,催化剂单程稳定性大于12 d的结果。催化剂的活性和稳定性与载体中的分子筛/Al2O3比例密切相关,Mo物种与载体的相互作用直接影响反应活性及催化剂寿命。经过多种表征手段证实,载体中添加Al2O3可以在一定程度上保护分子筛的骨架四配位Al,同时可以调节Mo物种的状态,使其在烯烃气氛下更易生成与歧化活性相关的Mo5+。聚合的烃类物质在Mo物种表面的沉积和Mo物种的深度还原是导致催化剂失活的主要原因[35]。
徐龙伢等开发的Mo/HMOR-Al2O3催化剂可以直接实现以1-丁烯为原料异构化-歧化制丙烯反应,通过催化剂优化可以达到较高的丙烯收率。通过调变催化剂酸性和反应条件可以调控反应诱导期长短和产物中丙烯/乙烯分布[63-65]。采用氨水蒸汽诱导法,将Al2O3涂层改性介孔SBA-15载体,实现了客体Al2O3在介孔孔壁上的均匀涂层(Al2O3@SBA-15),在保持开放介孔孔道的前提下,形成了利于Mo物种分散和低温歧化活性的Brönsted酸位。在温度120℃压力、0.1 MPa、MHSV=1.5 h-1的反应条件下,制备的Mo/Al2O3@SBA-15催化剂在1-丁烯歧化反应中表现出优异的歧化活性和反应稳定性[66]。
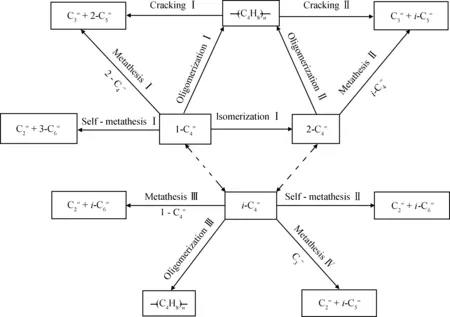
图3 混合碳四烯烃歧化反应路径网络[67]
3.2 长链烯烃歧化
目前,对长链烯烃歧化的研究主要以1-己烯、1-庚烯、1-辛烯、1-癸烯、十六碳烯等为研究对象,而且主要集中于催化基础研究,工艺研究较少。长链烯烃歧化工艺最具有代表性的是已经实现产业化的Shell Higher Olefin Process(SHOP)工艺。该工艺由Shell公司开发,由乙烯生产高碳线性α-烯烃和内烯烃,具体包括乙烯齐聚、α-烯烃异构化、内烯烃间歧化、产物循环等过程。其中,歧化单元采用MoO3/Al2O3催化剂,在100~125℃、1.0 MPa的反应条件下,C11~C14线性内烯烃的单程收率在10%~15%范围(质量分数)。最近,国内也有专利报道了一种用于乙烯直接与高碳烯烃(C11~C12)歧化制α-烯烃的负载型Re基催化剂的制备方法,克服了其它催化剂不能用于高碳烯烃歧化反应的缺陷[69]。
长链烯烃歧化也可以以液-固两相反应的形式发生。Fabris等[70]和Selva等[71-72]以超临界CO2为流动介质,考察了在连续流动条件下传统负载型Re2O3/Al2O3催化剂催化1-辛烯自歧化反应的活性。实验表明,与传统流动介质如甲苯、正己烷等相比,以超临界CO2作为流动介质,不仅避免了有毒溶剂的使用,同时能提高反应稳定性;尽管催化剂反应100~150 min后完全失活,但经过丙酮/超临界CO2原位再生后,在活性不降低的前提下,催化剂至少可以循环再利用5次。此外,与釜式反应方式相比,连续流动条件下更便于优化反应参数,但对压力比较敏感。实验得到最佳的反应温度和压力分别为100℃和9×106Pa,在1-辛烯和超临界CO2的流速分别为0.05和1 mL/min时,目的产物选择性高于90%。
4 多相催化剂催化烯烃歧化反应机理
早在20世纪中期,人们就发现了烯烃歧化反应现象。虽然相继提出了一些机理和反应过渡态[73-74],例如“环丁烷(Cyclobutane)”、“四亚甲基配
合物(Tetramethylene complex)”、“金属五元杂环(Metallacyclopentene)”等,但都不能完全解释观察到的实验现象。直到1971年,法国石油研究院(IFP)的Yves Chauvin教授和学生Jean-Louis Herisson在总结前人工作的基础上,首次提出了由金属卡宾活性物种[M=C]引发,经过“金属杂环丁烷(Metallacyclobutane)”中间体的“金属卡宾机理(Metal-alkylidene mechanism)”。目前,该机理已在均相歧化领域得到了证实和广泛应用,同时也在多相催化体系得到了普遍认可。
与均相催化剂不同,多相催化剂上一般不存在可以直接引发歧化反应的金属卡宾物种。关于初始金属卡宾物种是如何形成的问题成为困扰大家的一个难题,目前有以下几种解释。
(1) 烯烃分子首先吸附到配位不饱和的金属中心空轨道上形成π-complex,随后通过1,2-氢转移反应(以M-n-alkenyl为中间物)生成金属卡宾物种,如式(1)所示[75-76]。
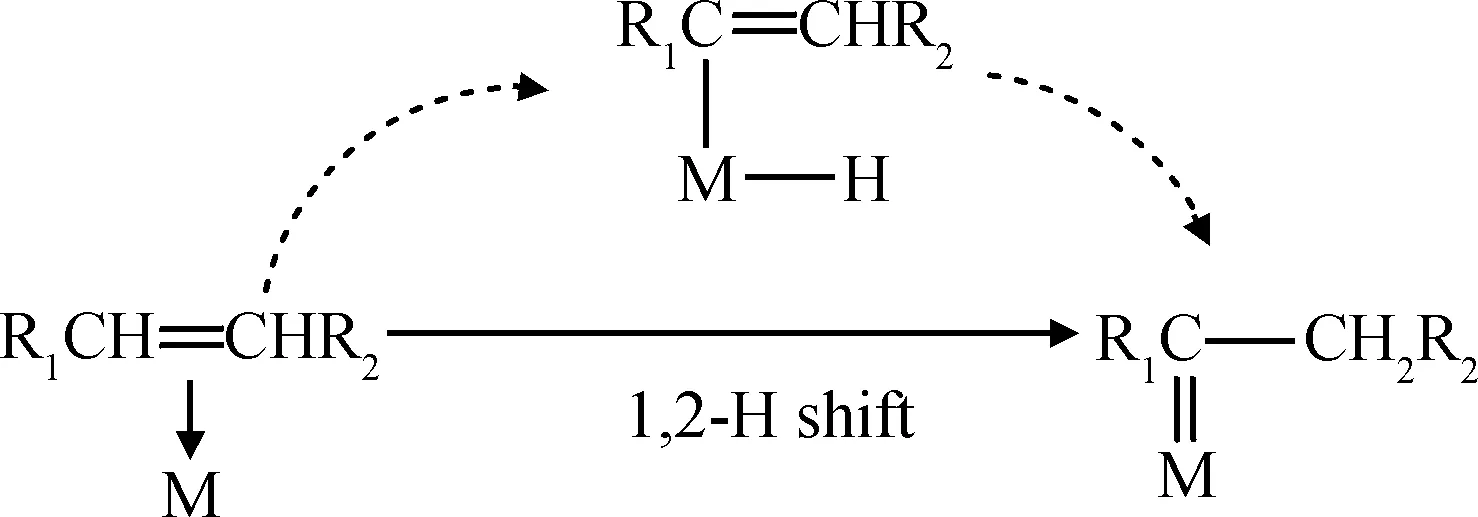
(1)
(2) 烯烃吸附到金属中心上之后,首先发生氢转移反应生成金属π配合物,经过金属四元杂环中间体生成初始金属卡宾物种,如式(2)所示。
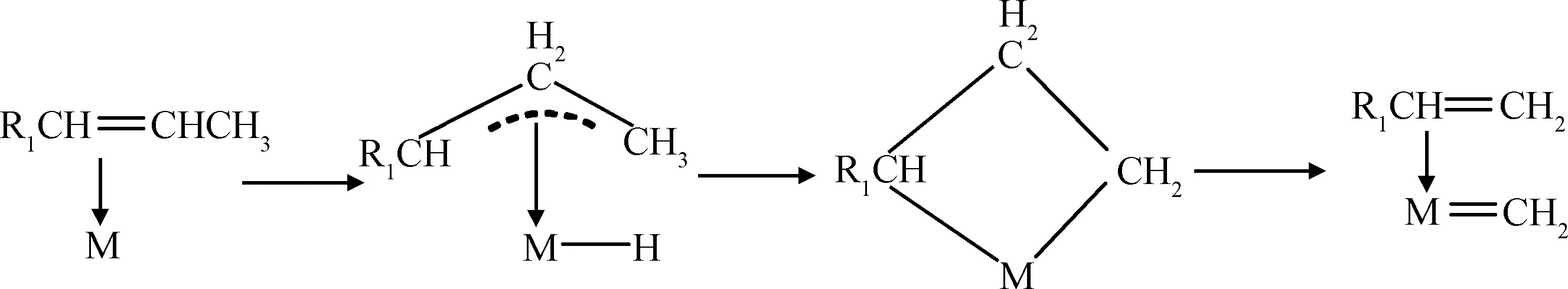
(2)
(3) 负载金属物种首先与载体表面羟基发生氧化加成反应生成金属氢化物,随后与反应物烯烃配位,并经过β-氢加成、α-氢消去反应生成初始卡宾物种,如式(3)所示[77-78]。
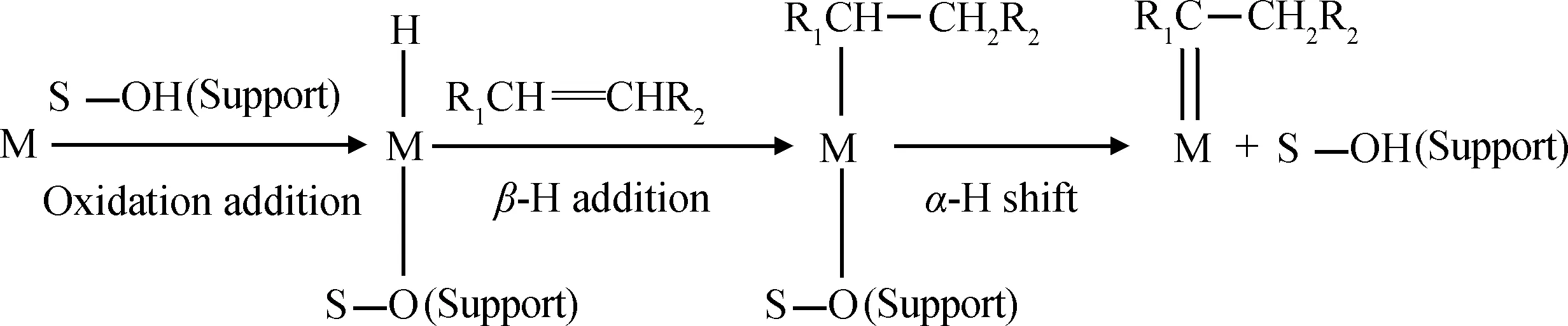
(3)
(4) 负载的金属氧化物的含氧基团首先与烯烃发生类Wittig反应,经过含氧四元环的中间物种最终生成金属卡宾和醛(酮)类物种,如式(4)所示[79-81]。某些研究者基于此模型,利用DFT理论计算初始金属卡宾物种的形成[42,82]。
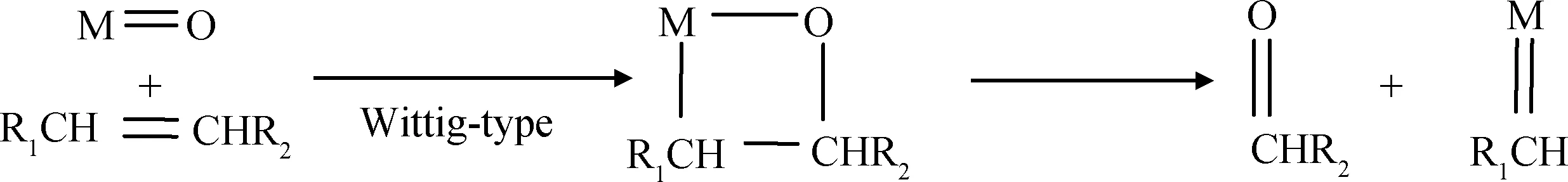
(4)
(5) 最近,Amakawa等[78]采用原位红外光谱和微量热法结合的方法考察了丙烯在Mo/SBA-15催化剂催化下自歧化反应生成初始卡宾物种的过程,并提出了一种初始卡宾形成的新路线。吸附的丙烯首先经催化剂表面上的酸性羟基(Brönsted酸位)质子化后生成异丙醇盐物种,随后被氧化脱氢为丙酮;丙酮脱附后,MoO3物种被还原至+4价,最后+4价的Mo物种与另一个丙烯分子经1,2-氢转移(氧化加成)反应生成初始活性卡宾物种,如式(5)所示,且与均相Schrock歧化催化剂具有类似的结构。
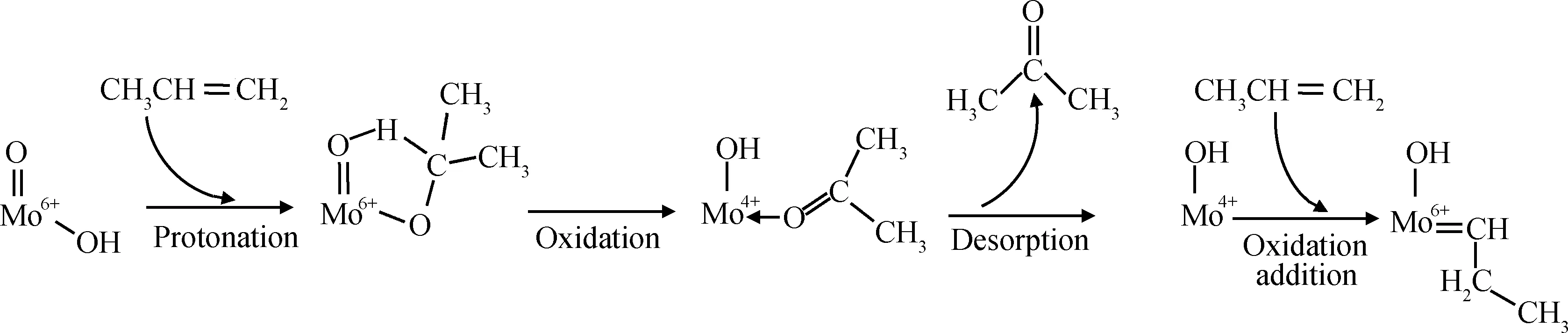
(5)
由于多相催化体系本身的不均一性及复杂性,卡宾物种难以直接量化分析。Handzlik等[83]和Amakawa等[78]均报道过负载型催化剂上测定活性金属卡宾中心数量的方法,以Amakawa等最新报道的方法为例予以说明。丙烯在Mo/SBA-15催化下自歧化反应生成乙烯和2-丁烯,反应初期存在诱导期,当反应达到稳态后对应催化剂上Mo卡宾数量达到最多。根据Chauvin教授提出的歧化反应机理,催化剂经过丙烯活化后会形成两种等量的卡宾物种Mo=CH2和Mo=CH-CH3。为此,可以用化学滴定法分析卡宾物种的数量[78],即当反应达到稳态后,用氘代乙烯CD2=CD2与卡宾物种Mo=CH-CH3反应生成的氘代丙烯CD2=CH-CH3(CD2=CD-CH3)的数量来反映催化剂上活性Mo物种的数量。此方法中,活性Mo物种的数量是生成的氘代丙烯CD2=CH-CH3的数量的2倍,如图4所示。实验证明,形成卡宾物种的数量不到总Mo物种数量的1%。
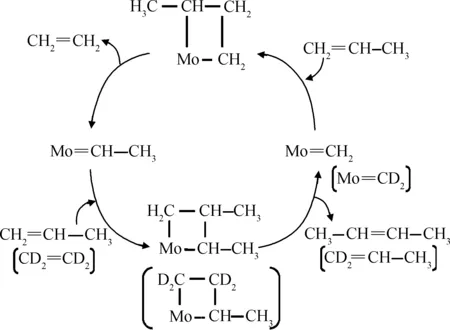
图4 丙烯歧化反应机理示意图[78]
5 结束语
与Re、W相比,Mo基催化剂在温和条件下即具有较高的歧化反应催化活性,而且存在工业化应用实例,具有广阔的发展前景。低碳烯烃歧化技术的关键是高效催化剂的开发,因此如何通过调变载体酸性、优化制备方法以及修饰金属物种来提高催化剂的寿命,减少副反应发生,成为多相Mo基烯烃歧化催化剂产业化面临的问题。
在碳四烯烃的歧化利用途径中,Mo基催化剂催化乙烯、丁烯歧化制丙烯反应是最具工业应用潜力的路线。在制备方法方面,普通浸渍与机械混合热分散法最易实现催化剂的规模放大。因为反应物烯烃分子的差异,同一催化剂催化不同烯烃分子的歧化活化中心会存在差异。在前期研究中,缺少采用原位光谱技术测定与表征反应过程中催化剂的活性位,因此对多相催化剂催化烯烃歧化反应机理的认识还有很大的发展空间。通过简单模型催化剂的制备,结合原位在线监测,实现催化剂结构与性能的关联,将对深化理解歧化反应机理和揭示催化剂的失活机制具有重要意义。
[1] BANKS R L, BAILEY G C. Olefin disproportionation: A new catalytic process[J]. I&EC Product Research Develop, 1964, 3(3): 170-173.
[2] BANKS R L, KUKES S G. New developments and concepts in enhancing activities of heterogeneous metathesis catalysts[J]. J Mol Catal, 1985, 28(1-3): 117-131.
[3] SCHROCK R R. Recent advances in high oxidation state Mo and W imido alkylidene chemistry[J]. Chem Rev, 2009, 109(8): 3211-3226.
[4] HOVEYDA A H, ZHUGRALIN A R. The remarkable metal-catalysed olefin metathesis reaction[J]. Nature, 2007, 450: 243-251.
[5] MOL J C. Olefin metathesis over supported rhenium oxide catalysts[J]. Catal Today, 1999, 51(2): 289-299.
[6] MOL J C. Industrial applications of olefin metathesis[J]. J Mol Catal A: Chem, 2004, 213(1): 39-45.
[7] SODESAWA T, OGATA E, KAMIYA Y. Disproportionation of propylene over MoO3-SiO2catalyst with various treatments[J]. Bull Chem Soc Jpn, 1979, 52(6): 1661-1664.
[8] AIGLER J M, HOUALLA M, HERCULES D M. Surface structure and metathesis activity of photoreducedallyl-based Mo/SiO2catalysts[J]. Top Catal, 2000, 10(1-2): 123-126.
[9] WANG Y D, CHEN Q L, YANG W M, et al. Effect of support nature on WO3/SiO2structure and butene-1 metathesis[J]. Appl Catal A, 2003, 250(1): 25-37.
[10] OIKAWA T, OOKOSHI T, TANAKA T, et al. A new heterogeneous olefin metathesis catalyst composed of rhenium oxide and mesoporous alumina[J]. Micropor Mesopor Mater, 2004, 74(1-3): 93-103.
[11] DEBECKER D P, MUTIN P H. Non-hydrolytic sol-gel routes to heterogeneous catalysts[J]. Chem Soc Rev, 2012, 41(9): 3624-3650.
[12] HUANG S J, LIU H J, ZHANG L, et al. Effects of acid leaching post-treatment on the catalytic performance of MoO3/mordenite-alumina catalysts for 1-butene metathesis reaction[J]. Appl Catal A, 2011, 404(1-2): 113-119.
[13] DEBECKER D P, STOYANOVA M, RODEMERCK U, et al. Thermal spreading as an alternative for the wet impregnation method: Advantages and downsides in the preparation of MoO3/SiO2-Al2O3metathesis catalysts[J]. J Phys Chem C, 2010, 114(43): 18664-18673.
[14] DEBECKER D P, STOYANOVA M, RODEMERCK U, et al. Facile preparation of MoO3/SiO2-Al2O3olefin metathesis catalysts by thermal spreading[J]. Stud Surf Sci, 2010, 175: 581-585.
[15] BALCAR H, HAMTIL R, ZILKOVA N, et al. Rhenium oxide supported on mesoporous organized alumina as a catalyst for metathesis of 1-alkenes[J]. Catal Lett, 2004, 97(1-2): 25-29.
[16] TOPKA P, BALCAR H, RATHOUSKY J, et al. Metathesis of 1-octene over MoO3supported on mesoporous molecular sieves: The influence of the support architecture[J]. Micropor Mesopor Mater, 2006, 96(1-3): 44-54.
[17] BALCAR H, CEJKA J. Green Metathesis Chemistry: Great Challenges in Synthesis[M]. Dordrecht: Springer, 2010: 101-114.
[18] HU J C, WANG Y D, CHEN L F, et al. Synthesis and characterization of tungsten-substituted SBA-15: An enhanced catalyst for 1-butene metathesis[J]. Micropor Mesopor Mater, 2006, 93(1-3): 158-163.
[19] 赵秦峰, 陈胜利, 高金森, 等. W-SBA-15的直接合成及其对2-丁烯与乙烯歧化制丙烯反应的催化性能[J]. 化工学报, 2009, 60(1): 75-82. (ZHAO Qinfeng, CHEN Shengli, GAO Jinsen, et al. Direct synthesis of W-SBA-15 and its catalytic performance in 2-butene and ethene metathesis to propene reaction[J]. Chin J Chem Ind Technol, 2009, 60(1): 75-82.)
[20] DEBECKER D P, HULEA V, MUTIN P H. Mesoporous mixed oxide catalysts via non-hydrolytic sol-gel: A review[J]. Appl Catal A, 2013, 451: 192-206.
[21] DEBECKER D P, BOUCHMELLA K, POLEUNIS C, et al. Design of MoO3-SiO2-Al2O3metathesis catalysts by nonhydrolytic sol-gel[J]. Chem Mater, 2009, 21(13): 2817-2824.
[22] DEBECKER D P, BOUCHMELLA K, STOYANOVA M, et al. A non-hydrolytic sol-gel route to highly active MoO3-SiO2-Al2O3metathesis catalysts[J]. Catal Sci Technol, 2012, 2(6): 1157-1164.
[23] DEBECKER D P, STOYANOVA M, COLBEAU-JUSTIN F, et al. One-pot aerosol route to MoO3-SiO2-Al2O3catalysts with ordered super microporosity and high olefin metathesis activity[J]. Angew Chem Int Ed, 2012, 51(9): 2129-2131.
[24] DEBECKER D P, STOYANOVA M, RODEMERCK U, et al. Aerosol route to nanostructured WO3-SiO2-Al2O3metathesis catalysts: Toward higher propene yield[J]. Appl Catal A, 2014, 470: 458-466.
[25] DEBECKER D P, SCHIMMOELLER B, STOYANOVA M, et al. Flame-made MoO3-SiO2-Al2O3metathesis catalysts with highly dispersed and highly active molybdate species[J]. J Catal, 2011, 277(2): 154-163.
[26] COPéRET C, CHABANAS M, PETROFF SAINT-ARROMAN R, et al. Homogeneous and heterogeneous catalysis: Bridging the gap through surface organometallic chemistry[J]. Angew Chem Int Ed, 2003, 42(2): 156-181.
[27] COPÉRET C. Design and understanding of heterogeneous alkene metathesis catalysts[J]. Dalton Trans, 2007, (47): 5498-5504.
[28] POATER A, SOLANS-MONFORT X, CLOT E, et al. Understanding d(0)-olefin metathesis catalysts: Which metal, which ligands?[J]. J Am Chem Soc, 2007, 129(26): 8207-8216.
[29] HANDZLIK J, OGONOWSKI J, STOCH J, et al. Properties and metathesis activity of molybdena-alumina, molybdena-silica-alumina and molybdena-silica catalysts- a comparative study[J]. Appl Catal A, 2006, 312: 213-219.
[30] HANDZLIK J, OGONOWSKI J, STOCH J, et al. Comparison of metathesis activity of catalysts prepared by anchoring of MoO2(acac)2on various supports[J]. Catal Lett, 2005, 101(1-2): 65-69.
[31] SIBEIJN M, MOL J C. Activity of supported Re2O7catalysts for the metathesis of methyl oleate[J]. Appl Catal, 1990, 67(2): 279-295.
[32] LI X J, ZHANG W P, LIU S L, et al. The role of alumina in the supported Mo/HBeta-Al2O3catalyst for olefin metathesis: A high-resolution solid-state NMR and electron microscopy study[J]. J Catal, 2007, 250(1): 55-66.
[33] LI X J, ZHANG W P, LIU S L, et al. A high-resolution MAS NMR study on the potential catalysts Mo/HBeta for olefin metathesis: The interaction of Mo species with HBeta zeolite[J]. J Mol Catal A-Chem, 2006, 250(1-2): 94-99.
[34] LI X J, ZHANG W P,LIU S L, et al. Olefin metathesis over heterogeneous catalysts: Interfacial interaction between Mo species and HBeta-Al2O3composite support[J]. J Phys Chem C, 2008, 112(15): 5955-5960.
[35] LI X J, ZHANG W P, LI X, et al. Insights into the deactivation mechanism of heterogeneous Mo/HBeta-Al2O3catalysts for olefin metathesis[J]. J Phys Chem C, 2009, 113(19): 8228-8233.
[36] LI X J, ZHANG W P, LIU S L, et al. Promoting effect of Mg in supported Mo/HBeta-Al2O3catalyst for cross-metathesis of ethene and butene-2 to propene[J]. J Mol Catal A-Chem, 2009, 313(1-2): 38-43.
[37] VANROOSMALEN A J, HARTMANN M C G, MOL J C. Active-centers for the isomerization of linear butenes on silica-gel-specific poisoning of bronsted acid sites by silylation[J]. J Catal, 1980, 66(1): 112-120.
[38] VANROOSMALEN A J, MOL J C. Active-centers for the metathesis and isomerization of alkenes on tungsten-oxide silica catalysts[J]. J Catal, 1982, 78(1): 17-23.
[39] MOSES A W, RAAB C, NELSON R C, et al. Spectroscopically distinct sites present in methyltrioxorhenium grafted onto silica-alumina, and their abilities to initiate olefin metathesis[J]. J Am Chem Soc, 2007, 129(28): 8912-8920.
[40] MOSES A W, RAMSAHYE N A, RAAB C, et al. Methyltrioxorhenium interactions with Lewis acid sites of an amorphous silica-alumina[J]. Organometallics, 2006, 25(9): 2157-2165.
[41] SCHEKLER-NAHAMA F, CLAUSE O, COMMEREUC D, et al. Influence of Lewis acidity of rhenium heptoxide supported on alumina catalyst on the catalytic performances in olefin metathesis[J]. Appl Catal A, 1998, 167(2): 237-245.
[42] LI X, ZHENG A M, GUAN J, et al. The effect of support acidity on olefin metathesis over heterogeneous Mo/HBeta catalyst: A DFT study[J]. Catal Lett, 2010, 138(1-2): 116-123.
[43] HANDZLIK J. Computational study of the properties and metathesis activity of Mo methylidene species in HZSM-5 zeolite[J]. J Mol Catal A, 2010, 316(1-2): 106-111.
[44] BALCAR H, CEJKA J. Mesoporous molecular sieves as advanced supports for olefin metathesis catalysts[J]. Coord Chem Rev, 2013, 257(21-22): 3107-3124.
[45] BALCAR H, HAMTIL R, ZILKOVA N, et al. Re(Ⅶ) oxide on mesoporous alumina of different types——Activity in the metathesis of olefins and their oxygen-containing derivatives[J]. Appl Catal A, 2007, 320: 56-63.
[46] SANG L, CHEN S L, YUAN G, et al. Metathesis of 1-butene and 2-butene to propene over Re2O7supported on macro-mesoporousγ-alumina prepared via a dual template method[J]. J Natur Gas Chem, 2012, 21(2): 105-108.
[47] OOKOSHI T, ONAKA M. A remarkable Mo catalyst for olefin metathesis: Hexagonal mesoporous silica-supported molybdenum oxide (MoO3/HMS)[J]. Chem Comm, 1998, (21): 2399-2400.
[48] BALCAR H, MISHRA D, MARCEAU E, et al. Molybdenum oxide catalysts for metathesis of higher 1-alkenes via supporting MoO2(acetylacetonate)2and MoO2(glycolate)2on SBA-15 mesoporous molecular sieves[J]. Appl Catal A, 2009, 359(1-2): 129-135.
[49] AGUADO J, ESCOLA J M, CASTRO M C, et al. Metathesis of 1-hexene over rhenium oxide supported on ordered mesoporous aluminas: comparison with Re2O7/gamma-Al2O3[J]. Appl Catal A, 2005, 284(1-2): 47-57.
[50] BAKALA P C, BRIOT E, MILLOT Y, et al. Comparison of olefin metathesis by rhenium-containing gamma-alumina or silica-aluminas and by some mesoporous analogues[J]. J Catal, 2008, 258(1): 61-70.
[51] HAMTIL R, ZILKOVA N, BALCAR H, et al. Rhenium oxide supported on organized mesoporous alumina——A highly active and versatile catalyst for alkene, diene, and cycloalkene metathesis[J]. Appl Catal A, 2006, 302(2): 193-200.
[52] ONAKA M, OIKAWA T. Olefin metathesis over mesoporous alumina-supported rhenium oxide catalyst[J]. Chem Lett, 2002, (8): 850-851.
[53] SANG L, CHEN S L, YUAN G, et al. Metathesis of 1-butene and 2-butene to propene over Re2O7supported on macro-mesoporousγ-alumina prepared via a dual template method[J]. J Natur Gas Chem, 2012, 21(2): 105-108.
[54] 宣东, 刘苏. 丁烯歧化生产己烯的方法: 中国, CN103030512[P]. 2011-09-30.
[55] NAKAMURA R, IIDA H, ECHIGOYA E. Synthesis of isoamylene from metathesis of isobutene with cis-2-butene[J]. Chem Lett, 1972, (4): 273-275.
[56] CHABANAS M, COPERET C, BASSET J M. Re-based heterogeneous catalysts for olefin metathesis prepared by surface organometallic chemistry: Reactivity and selectivity[J]. Chem Eur J, 2003, 9(4): 971-975.
[57] GARTSIDE R J, KLEINDIENST S R. Process and system for the production of isoprene: WO, 2011016842-A2[P]. 2011-02-10.
[58] 宣东, 王仰东, 刘苏. 异丁烯歧化制四甲基乙烯的方法: 中国, CN102372575[P]. 2010-08-23.
[59] AMAKAWA K, SUN L L, GUO C S, et al. How strain affects the reactivity of surface metal oxide catalysts[J]. Angew Chem Int Ed, 2013, 52(51): 13553-13557.
[60] LIU S L, HUANG S J, XIN W J, et al. Metathesis of ethylene and butylene-2 to propylene with Mo on HBeta-Al2O3catalysts[J]. Catal Today, 2004, 93(5): 471-476.
[61] LIU S L, LI X J, XIN W J, et al. Cross metathesis of butene-2 and ethene to propene over Mo/MCM-22-Al2O3catalysts with different Al2O3contents[J]. J Natur Gas Chem, 2010, 19(5): 482-486.
[62] ZHANG D Z, LI X J, LIU S L, et al. Effects of support acidity on the catalytic performance of Mo/HZSM-5-Al2O3catalysts in olefin metathesis[J]. Chin J Catal, 2011, 32(11): 1747-1754.
[63] LIU H J, HUANG S J, ZHANG L, et al. 1-Buene metathesis over a MoO3/H-mordenite-alumina catalyst[J]. Chin J Catal, 2008, 29(6): 513-518.
[64] LI X J, ZHANG D Z, ZHU X X, et al. 1-Butene metathesis over Mo/mordenite-alumina catalyst: Effect of sodium exchange degree in mordenite zeolite[J]. J Mol Catal A-Chem, 2013, 372: 121-127.
[65] LI X J, ZHU X X, ZHANG D Z, et al. 1-Butene isomerization and metathesis over Mo/mordenite-alumina: Factors influencing product distribution and induction period[J]. J Engergy Chem, 2013, 22(1): 145-150.
[66] 张大洲. 多相钼基催化剂上碳四烯烃歧化反应研究[D]. 大连: 中国科学院大连化学物理研究所, 2014.
[67] ZHANG D Z, LI X J, LIU S L, et al. Metathesis of C4 olefin over Mo-based heterogeneous catalysts: A novel route to propene and isopentene[J]. Appl Catal A: Gen, 2012, 439-440: 171-178.
[68] ZHANG D Z, LI X J, LIU S L, et al. Investigations into the C4 olefin metathesis over Mo/Al2O3: Effects of support nature and pretreatment conditions on the product distribution[J]. Appl Catal A: Gen, 2014, 472: 92-100.
[69] 郑来昌, 田志坚, 王如文,等. 一种用于高碳烯烃歧化制α-烯烃的负载型铼催化剂的制备方法: 中国, CN102794170A[P]. 2011-05-25.
[70] FABRIS M, AQUINO C, WARD A J, et al. Self-metathesis of 1-octene using alumina-supported Re2O7in supercritical CO2[J]. Top Catal, 2009, 52(3): 315-321.
[71] SELVA M, PEROSA A, FABRIS M, et al. The metathesis of alpha-olefins over supported Re-catalysts in supercritical CO2[J]. Green Chem, 2009, 11(2): 229-238.
[72] SELVA M, GUIDI S, PEROSA A, et al. Continuous-flow alkene metathesis: The model reaction of 1-octene catalyzed by Re2O7/gamma-Al2O3with supercritical CO2as a carrier[J]. Green Chem, 2012, 14(10): 2727-2737.
[73] CHAUVIN Y. Olefin metathesis: The early days[J]. Angew Chem Int Ed, 2006, 45(23): 3740-3747.
[74] ASTRUC D. The metathesis reactions: From a historical perspective to recent developments[J]. New J Chem, 2005, 29(1): 42-56.
[75] IWASAWA Y, HAMAMURA H. Mechanism for initial carbene formation in olefin metathesis over fixed Mo catalysts[J]. J Chem Soc-Chem Comm, 1983, 3: 130-132.
[76] IWASAWA Y, KUBO H, HAMAMURA H. Olefin metathesis over Al2O3-attached or SiO2-attached molybdenum catalysts - active structures and mechanism for initial carbene formation[J]. J Mol Catal, 1985, 28(1-3): 191-208.
[77] LAVERTY D T, ROONEY J J, STEWART A. Possible role of hydrido-metal complexes in metathesis, isomerization, dimerization, and polymerization of alkenes[J]. J Catal, 1976, 45(1): 110-113.
[78] AMAKAWA K, WRABETZ S, KROHNERT J, et al. In situ generation of active sites in olefin metathesis[J]. J Am Chem Soc, 2012, 134(28): 11462-11473.
[79] GRUNERT W, STAKHEEV A Y, FELDHAUS R, et al. Reduction and metathesis activity of MoO3/Al2O3catalysts: The activation of MoO3/Al2O3catalysts[J]. J Catal, 1992, 135(1): 287-299.
[80] BASRUR A G, PATWARDHAN S R, VYAS S N. Propene metathesis over silica-supported tungsten-oxide catalyst catalyst induction mechanism[J]. J Catal, 1991, 127(1): 86-95.
[81] SALAMEH A, COPERET C, BASSET J M, et al. Rhenium(VII) oxide/aluminum oxide: More experimental evidence for an oxametallacyclobutane intermediate and a pseudo-wittig initiation step in olefin metathesis[J]. Adv Synth Catal, 2007, 349(1-2): 238-242.
[82] GUAN J, YANG G, ZHOU D H, et al. The formation mechanism of Mo-methylidene species over Mo/HBeta catalysts for heterogeneous olefin metathesis: A density functional theory study[J]. J Mol Catal A, 2009, 300(1-2): 41-47.
[83] HANDZLIK J, OGONOWSKI J. Dynamic chemical counting of active centers of molybdena-alumina metathesis catalysts[J]. Catal Lett, 2003, 88(3-4): 119-122.
Olefin Metathesis Over Mo Based Heterogeneous Catalysts
LI Xiujie1, ZHANG Dazhou1,2, XIN Wenjie1, ZHU Xiangxue1, CHEN Fucun1, XIE Sujuan1, LIU Shenglin1, XU Longya1
(1.DalianInstituteofChemicalPhysics,ChineseAcademyofSciences,Dalian116023,China;2.WuhanEngineeringCo.Ltd,Wuhan430223,China)
Olefin metathesis, which is one of the most important C—C bond formation reactions, has
increasing attention. By the olefin metathesis the stocks of olefins upon the market demanding can be regulated at low energy and environmental cost which also provides a new way for the production of valued-added olefins. Heterogeneous catalysts for metathesis reactions are of further interest due to their ease of separation, good persistence and recyclability. This review focuses on the development of Mo based heterogeneous catalysts and its application in olefin metathesis reaction. Related new preparation methods, reaction types and working mechanism are summarized in detail.
olefin metathesis; heterogeneous catalysis; molybdenum; composite support
2014-10-27
国家自然科学基金项目(20903088, 21376235)资助
李秀杰,女,副研究员,博士,从事烯烃催化转化研究;Tel: 0411-84379279; E-mail:xiujieli@dicp.ac.cn
1001-8719(2015)02-0331-12
TQ426.96
A
10.3969/j.issn.1001-8719.2015.02.014
