DFT Study of H2Dissociation on MoxSyClusters
2015-06-21WangWeiZhaoXiaoguangLiHuifengZhouHanLiMingfeng
Wang Wei; Zhao Xiaoguang; Li Huifeng; Zhou Han; Li Mingfeng
(SINOPEC Research Institute of Petroleum Processing, Beijing 100083)
DFT Study of H2Dissociation on MoxSyClusters
Wang Wei; Zhao Xiaoguang; Li Huifeng; Zhou Han; Li Mingfeng
(SINOPEC Research Institute of Petroleum Processing, Beijing 100083)
A DFT study of H2dissociation on a series of MoxSyclusters was performed, including homolytic and heterolytic dissociation. The preference for the two pathways on these models show much difference, as the Mo coordination number increases, the homolytic dissociation becomes easier, whereas the heterolytic dissociation becomes more difficult. Furthermore, frontier orbital theory was used to analyze the dissociation mechanisms of these two pathways. It was found that the symmetry and energy gap of MoxSy’s HOMO and H2’s LUMO are the decisive factors in H2activation.
H2dissociation; MoS2; Frontier orbital theory
1 Introduction
Hydrogenation plays an important role in petroleum refining because it can upgrade the heavy oil and produce clean transportation fuels that meet the environmental regulations. The activation of molecular H2is the first step in the hydrogenation process, which not only generates active H but also impacts the structure of the active site and consequently impacts the hydrogenation (HYD) or hydrodesulphurization (HDS) reactions. Transition metal sulfides, especially molybdenum-based sulfides, have been widely used in the oil refining industry as hydrotreating catalysts; thus, H2activation on MoS2has been garnering significant attention[1]. A better understanding of the relationship between H2activation and the structure and properties of the active sites are of great interest to both the industry and the academia.
For decades, many spectroscopic[2-4], STM[5-6], H2—D2exchange[7], and TPD[8]studies have been performed on hydrogen activation. The experimental results indicated that H2activation usually occurs on the edges of MoS2, forming different types of -S-H and M-H species. The presence of the -SH group has been directly observed via inelastic neutron scattering, but there is no experimental evidence of Mo-H species. The mobility, acidity, and catalytic properties of the active H species were also investigated[4,7-9]. However, it is difficult to determine the nature of the active site and the mechanism of activation reaction using experimental methods.
In contrast, more detailed information has been obtained from theoretical studies. Thanks to the most likely equilibrium structure of the active sites under HDS conditions presented by STM, HRSTEM and the density-functional theory (DFT) calculations[10-14], many key issues have been investigated based on these possible sites, such as hydrogen adsorption, H2dissociation and migration pathways, the formation of S vacancies, the effect of promoting atoms Co and Ni, etc. For H2activation, two pathways were presented, as shown in Figure 1. One is homolytic dissociation on the S dimers to yield two -SH groups, and the other is the heterolytic dissociation on the S vacancy to yield -Mo-H and -S-H species[15]. H2activation through the two pathways on different sites have been widely studied by DFT calculations. Prodhomme, et al.[16]suggested that the preferred pathway for H2dissociative adsorption is heterolytic on M-edge of 50% S coverage and S-edge of 50% S coverage, and homolytic on S-edge of 100% S coverage and S-edge of 87% S coverage as S2dimers. Alexiev, et al.[17]suggested that hydrogen is adsorbed on the sulfur pairs but not on the Mo atoms on the surface. Cristol, et al.[14]investigated H2adsorption on the MoS2surfaces using periodical DFT studies, and the results showed that the hydrogen species are metastable on these surfaces with the least unstable configuration being the heterolytic dissociation of hydrogen on a metalsulfur pair. Travert, et al.[18]calculated the adsorption energies of H2on different unpromoted and promoted MoS2structures and suggested that the ability to dissociate H2depends on the nature of the metal atom and the sulfur coordination environment.

Figure 1 H2dissociation on MoS2: (1) homolytic dissociation; (2) heterolytic dissociation
These previous studies were usually performed using periodic or cluster models of crystalline MoS2, and the edge structures were always characterized by the S coverage (e.g., 0%, 50%, or 100%)[16], the coordination (e.g., MoIV, MoV, or MoVI)[18], or the geometry (e.g., Mo-edge, S-edge, or corner site)[19-21]. These concepts are actually the same. However, it is difficult to obtain a universal rule for H2dissociation based on these studies because they are usually based on the edge structure of MoS2crystals while partially considering several typical configurations of the active sites. Meanwhile, it is not convenient to compare the results with each other because different models and methods are used.
Consequently, a systematic study is needed to obtain some deep insight into the rules and nature of hydrogen activation. In contrast to the previous studies, we have designed a series of models and classified the active sites by the coordination numbers of the Mo and S atoms to solve this problem. In the following text, we first described the theoretical methods and the models we used in the present work. Then, the adsorption energy and activation barrier of the two pathways were calculated and compared in series. The results were briefly discussed and analyzed by using the frontier orbital theory. Finally, we determined the trend in the hydrogen activation ability as a function of the coordination number and noted the key factors of the H2activation catalyst.
2 Methods and Models
Computational results were obtained by using software programs provided by the Accelrys Software Inc. The DFT calculations were performed with the DMol3program[22-23], and graphical displays were generated by using Materials Studio. The physical wave functions were expanded in terms of accurate numerical basis sets, and DNP basis set were used. The generalized gradient approximation (GGA) functional developed by Perdew and Wang (PW91) was employed[24], and the medium quality mesh size was used for the numerical integration. The tolerances of energy, gradient, and displacement convergence were 0.000 02 Ha, 0.004 Ha/Å, and 0.005 Å, respectively. The real space cutoff of atomic orbital was 4.4 Å. The Linear Synchronous Transit/Quadratic Synchronous Transit method at the same level was used in searching for the transition states[25-26].
The models were clusters of Mo and S atoms with different coordinate states. In the MoS2clusters, Mo atom could be one- to six-fold coordinated by S atoms, and S atom could be one-, two-, or three-fold coordinated. Threefold coordinated S (SIII) is usually on the basal plane and considered to be inert toward H2activation[27], so only the one-fold and two-fold S atoms (SI, SII) are considered (the coordination number is represented using subscript). S atoms in the clusters that do not react with H2directly are saturated by H atoms. In these clusters, SIcould be considered as terminal S atoms on the edges. SIIcould be considered as the bridge S atoms. MoI-Vcould be considered as various unsaturated sites, while MoVIcould be considered as the saturated edge sites. By taking MoVISIand MoVISIIas the example, the model structures are shown in Figure 2.
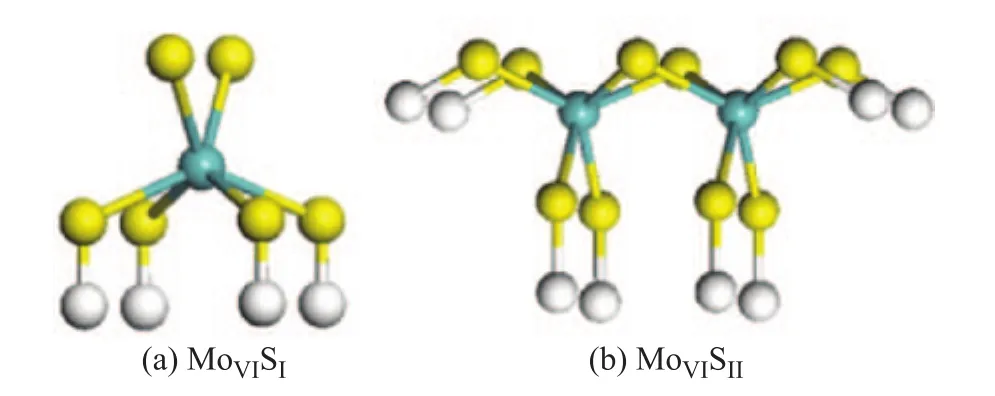
Figure 2 Models of MoxSyclusters
Consequently, a series of clusters composed of Mo (MoI, MoII, MoIII, MoIV, MoV, and MoVI) and S (SI, SII) were designed. Based on these clusters, the adsorption energies (Ead) and active barriers (Ea) during homolytic and heterolytic dissociation of H2were calculated. The adsorptionenergies were calculated by subtracting the energies of the optimized gas-phase H2and the cluster energy from the energy of the optimized H-H-cluster complex as shown in Eq. (1):

In addition, the purpose of this paper is to provide some insight into the nature of the mechanism during H2activation on MoS2by comparing a series of active sites, and a single calculation result is not meaningful unless it is compared with other results because the model is far from reality.
3 Results and Discussion
3.1 Adsorption energies and activation barriers
3.1.1 Homolytic dissociation
The adsorption energies and activation barriers of the homolytic dissociation of hydrogen are shown in Table 1.
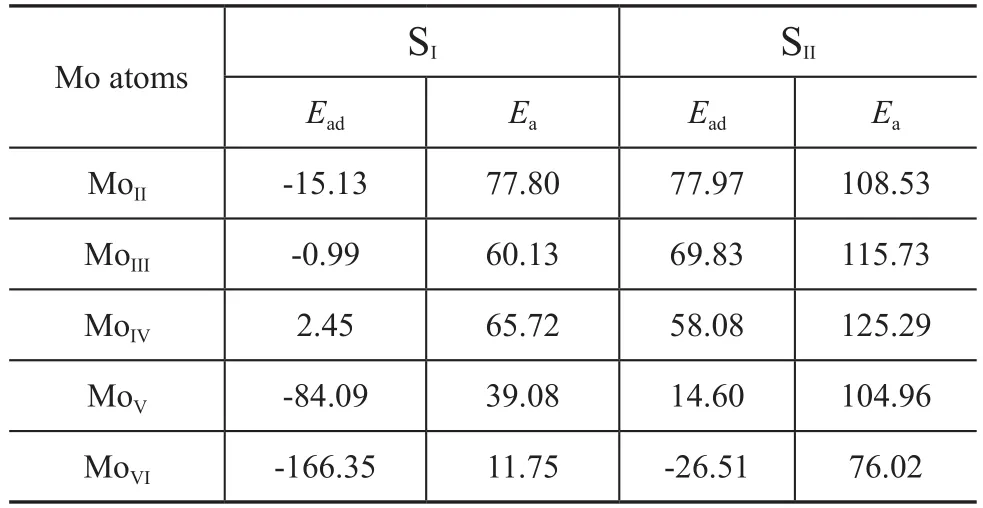
Table 1 Adsorption energies and activation barriers of the homolytic dissociation of H2kJ/mol
As for the homolytic dissociation on MoII-VISI, it can be concluded that both the adsorption energy and the activation barrier are negatively correlated with the Mo coordination number. It means that hydrogen dissociation on SI-SIbecomes easier if the Mo coordination number increases. As a result, hydrogen dissociation on MoVISIis the easiest in this series. This site is quite similar to the S2dimer on the 100% S coverage of Mo edge.
As for the homolytic dissociation on MoII-VISII, there is also a negative correlation between the adsorption energy and the Mo coordination number. However, the activation barrier first increases and then decreases as the Mo coordination number increases. As for MoVISII, this site is similar to S2on the 100%S coverage of S edge. The adsorption energy and activation barrier for this site are relatively small. Therefore, it should be relatively more active. But it is not so active as the S2dimer on the Mo edge.
3.1.2 Heterolytic dissociation
The adsorption energies and activation barriers of the heterolytic dissociation of H2are shown in Table 2.
As for the heterolytic dissociation of hydrogen on MoI-VISI, as shown in Table 2, there is no clear trend for the adsorption energy and activation barrier. The disordered variation may be ascribed to the reason that both the coordination number of the Mo and the structure of cluster will change when hydrogen heterolytically dissociates on MoI-VISI. We have also found out that the adsorption energy and activation barrier of H2on MoIVSIclusters are much larger than those of the other structures. It means that heterolytic dissociation of hydrogen on MoIVSIis more difficult, the reasons can be ascribed to that the 4d55s1electronic structure of the Mo contributes to more stable 4-fold and 6-fold coordinated Mo atoms.
As for the heterolytic dissociation on MoI-VISII, it could be concluded that both the hydrogen adsorption energy and the activation barrier are positively correlated with the Mo coordination number. It means that the heterolytic dissociation of H2becomes easier on MoI-VISIIas the Mo coordination number decreases.
Based on the former results, we can obtain the following conclusions. Firstly, the trends in the adsorption energy and activation barrier in each series are almost the same but are not identical. Secondly, the homolytic dissociation on two S atoms for both MoVISIand MoVISIIis easier. According to the work of Afanasiev, et al., S22-species located at edges are readily opened by hydrogen, even at relatively low temperatures, and they have a strong impact onthe catalytic function of MoS2-based catalysts[28-29]. Finally, the homolytic and heterolytic dissociations show quite different tendencies: the former becomes easier as the Mo coordination number increases, and the latter shows the opposite trend. To disclose the essential reasons for these different tendencies during hydrogen dissociation, the properties of the molecular orbitals and electrons during hydrogen activation were investigated in Section 3.2.

Table 2 Adsorption energies and activation barriers of the heterolytic dissociation of H2kJ/mol
3.2 Mechanism of H2dissociation
The frontier orbital theory has been used to analyze molecular interactions, such as adsorption or dissociation for decades[30-31]. This theory proposes that attractive interactions depend on the symmetry and relative energies of the HOMOs and LUMOs. Based on this point of view, molecular orbitals and ESP charges have been calculated and analyzed in detail in the following section. Because S atoms are in bridge S configurations (SII) under real hydrodesulphurization conditions based on recent DFT and STM studies[11,32], only dissociations on MoI-VISIIare considered as typical in this part.
3.2.1 Homolytic dissociation
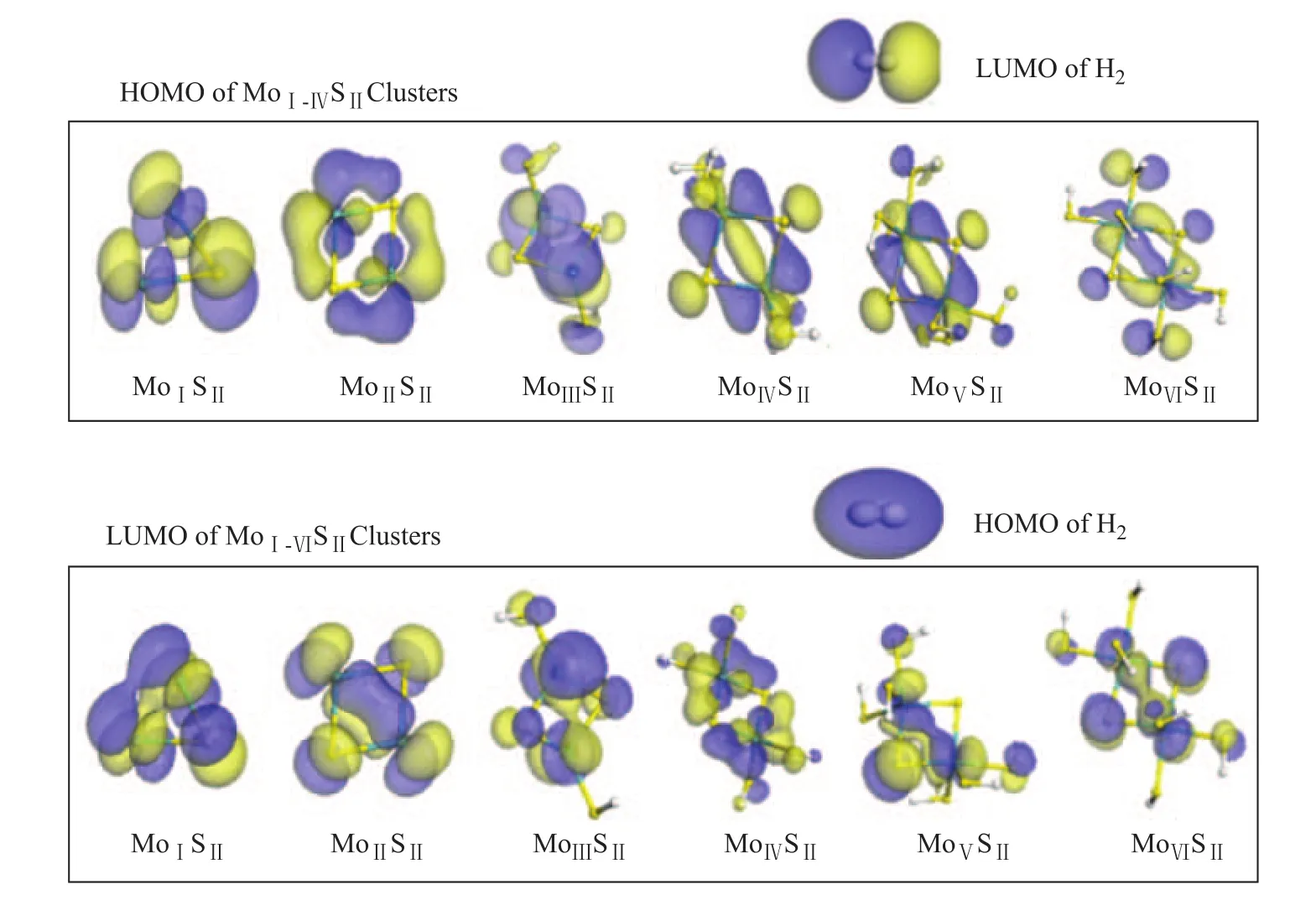
The symmetry matching of orbitals is crucial for a reaction to occur based on the frontier orbital theory. Therefore, we searched the frontier orbitals of the clusters firstly for the molecular orbitals over SII-SIIatoms that were compatible with hydrogen’s HOMO or LUMO. The molecular orbitals located around the HOMO-LUMO region were considered, including HOMO-3, HOMO-2, HOMO-1, HOMO, LUMO, LUMO+1, LUMO+2, and LUMO+3. The symmetry-adapted orbitals are shown in Figure 3. Then, the energy gaps between the cluster and H2were calculated and compared, as shown in Figure 4. Finally, the charge transfer during H2dissociation was analyzed using MoVISIIas an example.

Figure 3 Frontier orbitals of H2and MII-VISII
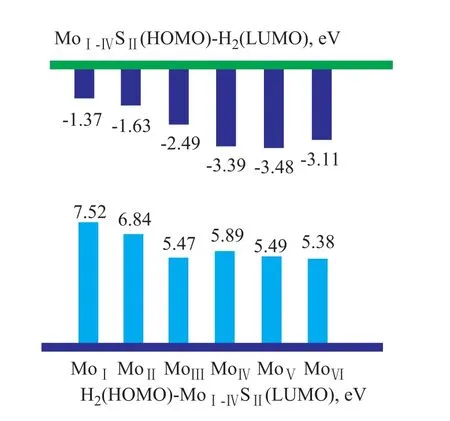
In Figure 4, the absolute values of the MoII-VISII(HOMO)-H2(LUMO) gap are in the range of between 3 eV to 5eV, which is smaller than the H2(HOMO)-MoII-VISII(LUMO) gap. In addition, the trend in the energy gaps between the MoII-VISII(HOMO) and the H2(LUMO) is quite similar to the trend in the activation barriers mentioned in Section 3.1.1, as shown in Figure 5. It suggests that the key factors for hydrogen activation are related with the MoII-VISIIHOMO and H2LUMO.
As shown in Figure 6, when H2approaches S2, the charge density on H2first increases then decreases until a transition state is formed. It suggests that electron density istransferred from the S2HOMO to the H2LUMO, through decreasing the strength of the H-H bonds, and finally breaking the bonds and forming two S-H groups. The S-H bond is a polar covalent bond, so H is relatively electropositive in the final state.
Sierraalta, et al.[33]studied the hydrogen interactions with S atoms from a topological point of view and suggested that H2is dissociated according to a four-center mechanism that involves two neighboring S atoms and a H2molecule. The mono-coordinated S (Smc) atoms are negatively charged, and the HOMO is localized over these atoms, suggesting that the charge transfer occurs from the Smcatoms to the H2anti-bonding ơ* orbital, which induces H2dissociation and makes the formation of the S-H bond easier[34], which is in agreement with our results.
3.2.2 Heterolytic dissociation
Similar to the previous studies, the MoI-VISIIorbitals that are compatible with hydrogen’s HOMO and LUMO were found and are shown in Figure 7. Then, the energy gaps between them were calculated, as shown in Figure 8.
In Figure 8, the absolute values of the MoI-VISII(HOMO)-H2(LUMO) gap are in the range of between 1 eV to 4 eV, which is smaller than the H2(HOMO)-MoI-VISII(LUMO) gap. In addition, the trend in the energy gaps between the MoI-VISII(HOMO) and the H2(LUMO) is quite similar to the trend in the activation barriers mentioned in Section 3.1.2, as shown in Figure 9. The results suggest that the heterolytic dissociation also depends on the MoI-VISIIHOMO and the H2LUMO.
As shown in Figure 10, the charge density of H2increases as the H2approaches MoVI-SII. Electron density is transferred from the S2HOMO to the H2LUMO, and the H-H bond is weakened and broken into S-H and Mo-H bonds. After the transition state is formed, the H atom that is closest to the S atom shows a positive charge, whereas the other H atoms (closest to Mo) show a negative charge, which may occur because the electronegativity order is Mo Some studies on metal-complexes have shown a mechanism for heterolytic dissociation of hydrogen on Mo-X[35-37]that is similar to the activation on metal. Firstly, H2molecules are adsorbed on Mn+, forming an η2-H2complex. Then, S2-transfers electron density to H—H ơ* to break the H—H bond. Finally, the H-H bond is cleaved heterolytically into Hδ+and Hδ-. However, the mechanism for heterolytic dissociation of hydrogen on MoS2was not clear until now. Based on the frontier orbital view, ourwork definitely indicates that the symmetry and energy gap of the HOMO of Mo and the LUMO of H2are the decisive factors in H2activation. Figure 4 Energy gaps between H2and MoII-VISII Figue 5Energy gaps and activation barriers of H2on MoII-VISI■—Energy gap;●—Activation barrier Figure 6 ESP charge of hydrogen during dissociation. Based on our conclusion, it could be expected that shifting the HOMO energy level of the Mo center may improve the catalytic performance of the catalyst. In addition, it should be noted that H2activation is just the first step of HDS or hydrogenation reactions, and in order to understand and control the entire reaction pathway, more other aspects should be considered, such as the properties ofthe active H, the adsorption of reactants, the reactions on the surface, and the effects of the geometry. And our work related to these issues is going on. Figure 7 Frontier orbitals of H2and MoI-VISII Figure 8 Energy gaps between H2and MoI-VISII Figure 9 Energy gaps and activation barriers of H2on MoI-VISII■—Energy gap;●—Activation barrier Figure 10 ESP charge of hydrogen during dissociation Both homolytic dissociation and heterolytic dissociation of H2were investigated on a series of clusters. Based on a comparison of the adsorption energies, activation barriers, and orbitals, two main conclusions were obtained. Firstly, with the increase in the Mo coordination number in connection with the homolytic dissociation of hydrogen, both the adsorption energy and activation barrier decrease, resulting in an increasing heterolytic dissociation of hydrogen. Secondly, the symmetry and the energy gap of MoxSy’s HOMO and hydrogen’s LUMO are the decisive factors during H2activation. Acknowledgements:The authors gratefully acknowledge the funding by the State Key Project (Grant Nos. 2012CB224802 and 2012BAE05B03) and by the Technology Development Project of SINOPEC (S112101). [1] Breysse M, Furimsky E, Kasztelan S, et al. Hydrogen activation by transition metal sulfides[J]. Catal Rev, 2002, 44(4): 651-735 [2] Wright C J, Sampson C, Fraser D, et al. Hydrogen sorption by molybdenum sulphide catalysts[J]. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 1980, 76(0): 1585-1598 [3] Sundberg P, Moyes R B, Tomkinson J. Inelastic neutron scattering spectroscopy of hydrogen adsorbed on powdered-MoS2, MoS2-alumina and nickel-promoted MoS2[J]. Bulletin des Sociétés Chimiques Belges, 1991, 100(11/12): 967-976 [4] Topsoe N Y, Topsoe H. FTIR studies of Mo/Al2O3-based catalysts: II. Evidence for the presence of SH groups and their role in acidity and activity[J]. J Catal, 1993, 139(2): 641-651 [5] Lauritsen J V, Nyberg M, Vang R T, et al. Chemistry of onedimensional metallic edge states in MoS2nanoclusters[J]. Nanotechnology. 2003, 14(3): 385 [6] Lauritsen J V, Nyberg M, Nørskov J K, et al. Hydrodesulfurization reaction pathways on MoS2nanoclusters revealed by scanning tunneling microscopy[J]. J Catal, 2004, 224(1): 94-106 [7] Thomas C, Vivier L, Lemberton J L, et al. Deuterium tracer studies on hydrotreating catalysts—isotopic exchange between hydrogen and hydrogen sulfide on sulfided NiMo/ Al2O3[J]. J Catal, 1997, 167(1): 1-11 [8] Jalowiecki L, Aboulaz A, Kasztelan S, et al. Hydrogenation and isomerization of alkadienes on powdered MoSxHy[J]. J Catal, 1989, 120(1): 108-117 [9] Wambeke A, Jalowiecki L, Kasztelan S, et al. The active site for isoprene hydrogenation on MoS2/γ-Al2O3catalysts[J]. J Catal, 1988, 109(2): 320-328 [10] Lauritsen J V, Bollinger M V, Lægsgaard E, et al. Atomicscale insight into structure and morphology changes of MoS2nanoclusters in hydrotreating catalysts[J]. J Catal, 2004, 221(2): 510-522 [11] Raybaud P, Hafner J, Kresse G, et al. Ab initio study of the H2–H2S/MoS2gas–solid interface: The nature of the catalytically active sites[J]. J Catal, 2000, 189(1): 129-146 [12] Raybaud P, Hafner J, Kresse G, et al. Structure, energetics, and electronic properties of the surface of a promoted MoS2catalyst: an ab initio local density functional study[J]. J Catal, 2000, 190(1): 128-143 [13] Hansen L P, Ramasse Q M, Kisielowski C, et al. Atomic-scale edge structures on industrial-style MoS2nanocatalysts[J]. Angewandte Chemie International Edition, 2011, 50(43): 10153-10156 [14] Cristol S, Paul J F, Payen E, et al. Theoretical study of the MoS2(100) surface: a chemical potential analysis of sulfur and hydrogen coverage[J]. J Phys Chem B, 2000, 104(47): 11220-11229 [15] Polz J, Zeilinger H, Müller B, et al. Hydrogen uptake by MoS2and sulfided alumina-supported Mo catalysts[J]. J Catal, 1989, 120(1): 22-28 [16] P rodhomme P, Raybaud P, Toulhoat H. Free-energy profiles along reduction pathways of MoS2M-edge and S-edge by dihydrogen: a first-principles study[J]. J Catal, 2011, 280(2): 178-195 [17] Alexiev V, Prins R, Weber T. DFT study of MoS2and hydrogen adsorbed on the (10-10) face of MoS2[J]. Phys Chem Chem Phys. 2001, 3: 5326-5336 [18] Travert A, Nakamura H, van Santen R A, et al. Hydrogen activation on Mo-based sulfide catalysts, a periodic DFT study[J]. J Am Chem Soc, 2002, 124(24): 7084-7095 [19] Sun M, Nelson A E, Adjaye J. Ab initio DFT study of hy-drogen dissociation on MoS2, NiMoS, and CoMoS: Mechanism, kinetics, and vibrational frequencies[J]. J Catal, 2005, 233(2): 411-421 [20] Sun M, Nelson A E, Adjaye J. Adsorption and dissociation of H2and H2S on MoS2and NiMoS catalysts[J]. Catal Today, 2005, 105(1): 36-43 [21] Wen X, Zeng T, Teng B, et al. Hydrogen adsorption on a Mo27S54cluster: a density functional theory study[J]. J Mol Catal A: Chem, 2006, 249(1/2): 191-200 [22] Delley B. From molecules to solids with the DMol3approach[J]. J Chem Phys, 2000, 113(18): 7756-7764 [23] Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Phys Rev, 1965, 140(4A): A1133-A1138 [24] Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Phys Rev B, 1992, 45(23): 13244-13249 [25] Bell S, Crighton J S. Locating transition states[J]. J Chem Phys, 1984, 80(6): 2464-2475 [26] Fischer S, Karplus M. Conjugate peak refinement: an algorithm for finding reaction paths and accurate transition states in systems with many degrees of freedom[J]. Chem Phys Lett, 1992, 194(3): 252-261 [27] Wiegenstein C G, Schulz K H. Deuterium and deuterium sulfide adsorption on MoS2(001)[J]. Surf Sci, 1998, 396(1/3): 284-294 [28] A fanasiev P, Jobic H, Lorentz C, et al. Low-temperature hydrogen interaction with amorphous molybdenum sulfides MoSx[J]. J Phys Chem C, 2009, 113(10): 4139-4146 [29] A fanasiev P. The influence of reducing and sulfiding conditions on the properties of unsupported MoS2-based catalysts[J]. J Catal, 2010, 269(2): 269-280 [30] Tong Y Y, Mériaudeau P, Renouprez A J, et al. Tailoring the frontier orbitals at the surfaces of platinum catalysts[J]. J Phys Chem B, 1997, 101(49): 10155-10158 [31] Hoffmann R. A chemical and theoretical way to look at bonding on surfaces[J]. Rev Mod Phys, 1988, 60(3): 601-628 [32] Lauritsen J V, Bollinger M V, Lægsgaard E, et al. Atomicscale insight into structure and morphology changes of MoS2nanoclusters in hydrotreating catalysts[J]. J Catal, 2004, 221(2): 510-522 [33] Sierraalta A, Ruette F. H2interaction with S atoms of a MoS2modelled catalytic site: electronic density analysis for SH formation[J]. J Mol Catal A: Chem, 1996, 109(3): 227-238 [34] Lobos S, Sierraalta A, Ruette F, et al. Modeling MoS2catalytic surface with simple clusters[J]. J Mol Catal A: Chem, 2003, 192(1–2): 203-216 [35] Kubas G J. Hydrogen activation on organometallic complexes and H2production, utilization, and storage for future energy[J]. J Organomet Chem, 2009, 694(17): 2648-2653 [36] Könczöl L, Makkos E, Bourissou D, et al. Computational evidence for a new type of η2-H2complex: when main-group elements act in concert to emulate transition metals[J]. Angewandte Chemie, 2012, 124(38): 9659-9662 [37] Kubas G J. Metal–dihydrogen and ơ-bond coordination: the consummate extension of the Dewar–Chatt–Duncanson model for metal–olefin π bonding[J]. J Organomet Chem, 2001, 635(1/2): 37-68 The Project “Study on Technology for Manufacture of High-Spheroidicity FCC Catalysts” Passed SINOPEC’s Appraisal On January 7, 2015 the project “Study on the technology for manufacture of high-spheroidicity FCC catalysts “undertaken by the SINOPEC Research Institute of Petroleum Processing (RIPP) has passed the technical appraisal organized by the Science and Technology Division of the Sinopec Corp. The research team engaging in the development of the said technology has designed a method for characterization of spheroidicity of FCC catalysts and put forward the definition of catalyst spheroidicity index, as well as the empirical formula for calculating the spheroidicity of FCC catalysts. The outcome of commercial application of this technology for preparing the high-spheroidicity FCC catalysts after its emergence has revealed that the spheroidicity of FCC catalyst has increased by 0.023 at the Qilu Branch of SINOPEC Catalyst Company, with the catalyst spheroidicity index increased by a value of 46. date: 2014-08-19; Accepted date: 2014-11-09. Zhao Xiaoguang, Telephone: +86-10-82368080; E-mail: zhaoxg.ripp@sinopec.com.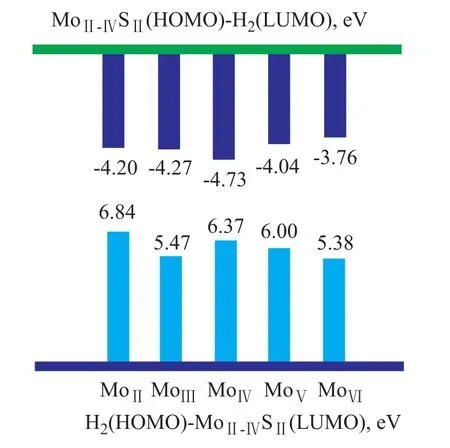




4 Conclusions
杂志排行

中国炼油与石油化工的其它文章
- Study on Surface Adsorption and Inhibition Behavior of Corrosion Inhibitors Contained in Copper Foil Rolling Oil
- Synthesis of Petroleum Sulfonate Surfactant with Ultra-Low Interfacial Tension in Rotating Packed Bed Reactor
- Simultaneous Removal of H2S and Organosulfur Compounds from Lique fied Petroleum Gas Using Formulated Solvents: Solubility Parameter Investigation and Industrial Test
- Effect of Dodecylbenzene Sulfonic Acid Used as Additive on Residue Hydrotreating
- Sulfur Distribution during Hydrothermal Liquefaction of Lignite, Wheat Straw and Plastic Waste in Sub-Critical Water
- Simulation Optimization and Experimental Study of Cross-Wall Adiabatic Dividing Wall Column Used to Separate Hexane-Heptane-Octane System