壮药黄花调气饮的质量控制*
2015-06-15黄蓓刘华钢梁美艳朱丹黄敏
黄蓓,刘华钢,梁美艳,朱丹,黄敏
壮药黄花调气饮的质量控制*
黄蓓1,刘华钢2,梁美艳3,朱丹2,黄敏1
(1.广西中医药大学附属瑞康医院药物研发中心,南宁 530011;2.广西医科大学药学院,南宁 530021;3.广西壮族自治区梧州市食品药品检验所,梧州 543000)
目的 建立壮药黄花调气饮的质量标准。方法 采用薄层色谱法对黄芪、山银花、甜茶进行定性鉴别;采用高效液相色谱法测定山银花中绿原酸含量,Ultimate色谱柱(250 mm×4.6 mm,5 μm),甲醇-水-冰醋酸(20:80:1)为流动相,柱温30 ℃,流速1.0 mL·min-1,检测波长327 nm,进样量10 μL。结果 黄芪、山银花、甜茶薄层色谱斑点清晰,专属性强。绿原酸在0.141 6~0.991 2 μg范围内呈良好的线性关系(r=0.999 9),平均回收率为102.2%,RSD为0.68%。结论 该方法简便易行,重复性较好,可用于壮药黄花调气饮的质量控制。
壮药;黄花调气饮;色谱法,薄层;色谱法,高效液相
壮药黄花调气饮为刘华钢教授根据民间验方,运用壮医独特的“调气机,通三道”治疗原则和临床诊疗经验研制而成的茶剂,该制剂现正在进行制剂注册申请。制剂处方由黄芪、山银花、大枣、浮小麦、甜茶组成,生产工艺为取黄芪、浮小麦、大枣加水煎煮2次,合并煎液,滤过,滤液浓缩成清膏后,加入山银花、甜茶粗粉,烘干,粉碎成粗粉,制成袋泡茶。其具有顺气解毒、调气补虚的功用,可通调气道,顺气解毒,用于四时感冒,尤以虚人感冒、老人感冒、小儿感冒更宜[1-3]。为了控制制剂质量,对处方中黄芪、山银花、甜茶进行薄层色谱(thin layer chromatography,TLC)法鉴别,并采用高效液相色谱(high performance liquid chromatography,HPLC)法测定制剂中山银花[4]的有效成分绿原酸[5-6]。所建立的方法具有较好的分离度、重复性、专属性,且操作简便准确,可以有效地控制壮药黄花调气饮的质量。
1 仪器与试药
1.1 仪器 岛津高效液相色谱仪(包括LC-20A元泵,SIL-20A标准自动进样器,SPD-20A紫外-可见检测器,CTO-20A柱温箱,CBM-20A工作站);Sartorius BP211D电子天平(赛多利斯科学仪器有限公司);SK2200LHC超声波清洗机(上海科导超声仪器有限公司);Agilent8453紫外-可见分光光度计(美国安捷伦);LG16-W高速离心机(北京医用离心机厂);硅胶G板、聚酰胺薄膜(浙江省台州市路桥四甲生化塑料厂)。
1.2 试药 黄芪对照药材(中国食品药品检定研究院,批号:20120502);绿原酸对照品(中国食品药品检定研究院,批号:110753-200413);甜茶对照药材(南宁生源中药饮片有限责任公司,经广西中医药大学韦松基教授鉴定为蔷薇科植物甜叶悬钩子RubussuavissimusS.Lee的干燥叶,批号:100201);甲醇(色谱纯,美国 Fisher 公司);水为超纯水,其余试剂为分析纯。壮药黄花调气饮(批号:20110605,20110610,20110614)由广西中医药大学附属瑞康医院制剂室生产。
2 TLC鉴别
2.1 山银花 取本品0.5 g,加甲醇10 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取缺山银花的阴性对照样品0.5 g,同法制成阴性对照溶液。再取绿原酸对照品,加甲醇制成每毫升含1 mg的溶液,作为对照品溶液。照薄层色谱法[7]实验,吸取上述溶液各2 μL分别点于同一聚酰胺薄膜上,以36%乙酸为展开剂,展开,取出,晾干,置紫外灯光(波长365 nm)下检视。供试品色谱中,在与对照品色谱相应位置上,显相同颜色的荧光斑点。缺山银花阴性样品无相应斑点。见图1。

1~3.供试品;4.绿原酸;5.阴性对照
2.2 黄芪 取本品3.5 g,加甲醇30 mL,超声处理1 h,滤过,滤液加于中性氧化铝柱(100~200目,10 g,内径为10 mm)上,用40%甲醇100 mL洗脱,收集洗脱液,蒸干,残渣加水30 mL使溶解,用水饱和的正丁醇振摇提取2次,每次20 mL,合并正丁醇液,用水洗2次,每次20 mL,弃去水液,正丁醇液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取缺黄芪的阴性对照样品3.5 g,同法制成阴性对照溶液。再取黄芪对照药材3 g,加甲醇30 mL,超声处理1 h,滤过,滤液加于中性氧化铝柱(100~200目,5 g,内径为10 mm)上,从“用40%甲醇100 mL洗脱”起,同法制成对照药材溶液。照薄层色谱法实验,吸取上述溶液各5~10 μL分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯(5:5)为展开剂,展开,取出,晾干,置氨蒸气中熏后,置紫外灯光(波长365 nm)下检视。供试品色谱中,在与对照药材色谱相应位置上,显相同颜色的荧光主斑点。见图2。
2.3 甜茶 取本品1.0 g,加甲醇10 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取缺甜茶的阴性对照样品1.0 g,同法制成阴性对照溶液。再取甜茶对照药材0.5 g,同法制成对照药材溶液,照薄层色谱法实验,吸取上述溶液各5~10 μL分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯-甲酸(8:2:0.4)为展开剂,展开,取出,晾干,置紫外灯光(波长365 nm)下检视。供试品色谱中,在与对照药材色谱相应位置上,显相同颜色的荧光主斑点。见图3。
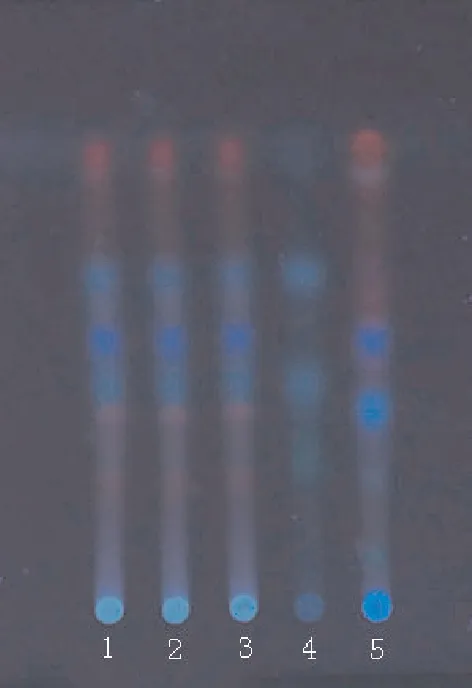
1~3.供试品;4.黄芪对照药材;5.阴性对照
1-3.sample;4.Astragaliradixreference ;5.negative control
Fig.2 TLC chromatogram ofAstragaliradix
1-3.sample;4.Foliumrubisuavissimireference;5.negative control
Fig.3 TLC chromatogram ofFoliumrubisuavissimi
3 HPLC法测定山银花中绿原酸
3.1 色谱条件 Ultimate色谱柱(250 mm×4.6 mm,5 μm);用十八烷基硅烷键合硅胶为填充剂;以甲醇-水-冰醋酸(20:80:1)为流动相;柱温30 ℃;检测波长327 nm;流速1.0 mL·min-1;进样量10 μL[8-9]。
3.2 对照品溶液的制备 精密称取绿原酸对照品适量,制成每毫升含绿原酸70 μg 。
3.3 供试品溶液的制备 取本品约1 g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25 mL,密塞,称定质量,超声处理30 min,放冷,再称定质量,用50%甲醇补足减失的质量,摇匀,滤过,精密吸取续滤液5 mL,置25 mL棕色瓶中,加50%甲醇至刻度,摇匀,滤过,取续滤液,即得。
3.4 阴性样品溶液的制备 分别按处方比例及制剂制备工艺制备不含山银花的阴性样品,再按“3.3”项下方法制备,即得。
3.5 专属性实验 取阴性样品溶液、对照品溶液和样品溶液,按照“3.1”项下色谱条件,注入高效液相色谱仪,进样量10 μL,阴性溶液在绿原酸色谱峰处无干扰峰出现,表明对被测成分无干扰。理论板数按绿原酸峰计算应不低于6 000。见图4。
3.6 线性关系考察 精密称取绿原酸对照品1.77 mg置于25 mL量瓶中,加甲醇溶解并定容,制成浓度为70.8 μg·mL-1的绿原酸对照品溶液,取上述绿原酸对照品溶液分别进样2,4,6,8,10,12,14 μL记录绿原酸峰面积值。以对照品进样量(μg)为横坐标(X),对应峰面积为纵坐标(Y),绘制标准曲线,得回归方程Y=3.2×106X-22 423(r=0.999 9),结果表明绿原酸进样量在0.141 6~0.991 2 μg范围内与峰面积呈良好线性关系。
3.7 精密度实验 取同一供试品溶液,按“3.1”项下色谱条件,连续进样6次,进行分析测定绿原酸峰面积值,计算绿原酸含量RSD值为0.35%。
3.8 稳定性实验 取同一供试品溶液,分别在0,2,4,6,8,10,12 h进样,进样量为10 μL,分别测定其峰面积值,RSD=0.37%(n=7),表明供试品溶液在12 h内稳定,
3.9 重复性实验 取同一供试品,按“3.3”项下方法重复提取6份作为供试品溶液,按“3.1”项色谱条件进行测定,记录绿原酸峰面积,求出其含量,计算RSD值为0.25%(n=6)。
3.10 加样回收率实验 精密称取己知含量的样品6份,每份约0.5 g,分别精密加入4.29 mg·mL-1绿原酸对照品溶液1 mL,按“3.3”项下方法制备供试品,按照“3.1”项下色谱条件分别进样,进样量10 μL,测定峰面积,绿原酸的加样回收率为102.2%,RSD为0.68%。见表1。
3.11 样品测定 按上述色谱条件测定3批制剂中绿原酸含量,每批制剂测3份,平均含量分别为8.01,7.90,7.92 mg·g-1。
4 讨论
笔者曾对制剂中另二味药大枣、浮小麦进行过薄层鉴别[10],结果在与对照药材色谱相应位置上,有相同颜色的荧光斑点,但阴性对照存在干扰,大枣和浮小麦的薄层鉴别不宜作为壮药黄花调气饮的薄层鉴别项目。
根据文献报道,绿原酸对照品溶液在327 nm波长处有最大吸收[11-14]。取绿原酸对照品的甲醇溶液,在紫外分光光度计上进行扫描,测得最大吸收波长为327 nm,与文献报道相吻合,故本实验采用327 nm为检测波长。
笔者曾试用流动相:乙腈-0.4%磷酸溶液(13:87)[15],甲醇-水-冰醋酸(15:85:1),甲醇-水-冰醋酸(25:75:1),甲醇-水-冰醋酸(20:80:1)。实验条件摸索证实,采用流动相甲醇-水-冰醋酸(20:80:1)时,样品中绿原酸与其他杂质峰分离较好,并能在15 min内出峰完毕,达到分析要求,故选择甲醇-水-冰醋酸(20:80:1)为流动相。
笔者比较25,30,35 ℃柱温对高效液相色谱的影响,实验发现,柱温对绿原酸峰的分离度和保留时间影响不大,因此选择较接近室温的柱温30 ℃。
[1] 朱丹,刘华钢,黄慧学.金黄Ⅰ号及组方药味体外抗流感病毒作用[J].中国实验方剂学杂志,2010,16(17):181-183.
[2] 刘华钢,朱丹.金黄Ⅰ号对小鼠免疫功能的影响[J].中国中药杂志,2007,32(11):1116-1117.
[3] 朱丹,张春花,梁秋云,等.金黄扶正茶对免疫抑制小鼠细胞免疫功能的影响[J].中国实验方剂学杂志,2012,18(8):242-245.
[4] 广西壮族自治区食品药品监督管理局.广西壮族自治区壮药质量标准:2008年版[M].南宁:广西科学技术出版社,2008:39.
[5] 卢凤来,蒋海英,陈月圆,等.HPLC-ELSD法测定山银花中绿原酸、灰毡毛忍冬皂苷乙和川续断皂苷乙[J].中成药,2013,35(8):1821-1823.
[6] 李锦燊,吴洪文.HPLC同时测定山银花中绿原酸和木犀草苷的含量[J].北方药学,2013,10(8):10-11.
[7] 国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:附录34.
[8] 李艳丽,许亮,杨燕云,等.HPLC测定不同产地牛蒡草中绿原酸含量[J].中国实验方剂学杂志,2013,18(23):117-119.
[9] 孙艳涛,赵兰英,李婷婷,等.高效液相色谱切换波长法同时测定牛蒡子中活性成分的含量[J].医药导报,2014,33(1):100-102.
[10] 孟霜,李慧峰,闫艳,等.浮小麦药材质量控制研究[J].中国实验方剂学杂志,2012,18(24):124-127.
[11] 陈黎,王启斌,朱海涛,等.HPLC法测定风湿Ⅱ号合剂中绿原酸的含量[J].中国药房,2010,21(31):2920-2921.
[12] 李玲,刘贻珍.HPLC法测定咽炎合剂中绿原酸的含量[J].中国药事,2012,26(12):1372-1373.
[13] 周燕园.HPLC法测定墨旱莲配方颗粒中绿原酸和咖啡酸[J].中成药,2012,34(2):374-377.
[14] 徐岳鑫.HPLC法测定青果丸中绿原酸的含量[J].中国药师,2013,16(2):255,304.
[15] 纪国力,刘玉玲.HPLC法测定羚翘解毒丸中绿原酸的含量[J].中国医药科学,2012,2(24):109-110.
Quality Control ofHuanghuaTiaoqiDecoction ofZhuangMedicine
HUANG Bei1, LIU Huagang2, LIANG Meiyan3, ZHU Dan2, HUANG min1
(1.RuikangHospitalAffiliatedtoGuangxiUniversityofChineseMedicine,GuangxiZhuangAutonomousRegion,Nanning530011,China; 2.GuangxiMedicalUniversity,Nanning530021,China; 3.WuzhouFoodandDrugAdministration,GuangxiZhuangAutonomousRegion,Wuzhou543000,China)
Objective To develop a quality control standard ofhuanghuatiaoqidecoction ofZhuangMedicine. Methods Thin layer chromatography (TLC) was applied to identifyAstragaliradix,Loniceraeflos, andFoliumrubisuavissimi.HPLC was employed to determine the content of chlorogenic acid inLoniceraeflos.The chromatography conditions consisted of Ultimate column (250 mm×4.6 mm, 5 μm) with mobile phase of methaonl-water-glacial acetic acid (20:80:1), column temperature of 30 ℃, flow rate of 1.0 mL·min-1, UV detection wavelength of 327 nm, and injection volume of 10 μL. ResultsAstragaliradix,Loniceraeflos,Foliumrubisuavissimiwere indentified by TLC.Chlorogenic acid showed a good linear relationship at a range of 0.141 6-0.991 2 μg,r=0.999 9.The average recovery was 102.2%, and RSD was 0.68%. Conclusion The methods are shown to simple to operate, and can be used for the quality control ofhuanghuatiaoqidecoction ofZhuangMedicine.
ZhuangMedicine;Huanghuatiaoqidecoction; Chromatography, thin layer; Chromatography, high performance liquid
2014-02-25
2014-06-06
*广西中医药管理局项目(GZYZ-10-31);广西教育厅项目(201010LX051);广西科学研究与技术开发计划项目(桂科攻0332007)
黄蓓(1975-),女,广西南宁人,副主任中药师,学士,从事新药研发工作。电话:(0)13217718960,E-mail:pp0425pp@163.com。
刘华钢(1956-),女,教授,博士生导师,博士,主要从事药理学及药剂学研究。电话:0771-5700208,E-mail:hgliu206@263.net。
R286;R927.2
B
1004-0781(2015)02-0252-04
DOI 10.3870/yydb.2015.02.030
