笠贝幼虫变态相关基因筛选及分子网络构建
2015-06-09张丽莉王国栋王艺磊
尹 诚,张丽莉,王国栋,王艺磊
(1.集美大学水产学院,福建厦门 361021;2.农业部东海海水健康重点实验室,福建厦门 361021)
笠贝幼虫变态相关基因筛选及分子网络构建
尹 诚1,2,张丽莉1,2,王国栋1,2,王艺磊1,2
(1.集美大学水产学院,福建厦门 361021;2.农业部东海海水健康重点实验室,福建厦门 361021)
利用NCBI在线数字化差异显示工具(Digital Differential Display)对笠贝(Lottiagigantea)EST数据库进行筛选,获得笠贝幼虫时期显著差异表达基因203个,其中124个基因有Blast2Go注释结果,这124个基因可以归类成细胞组分、分子功能和生物学过程三大类;参与MAPK、Ras、PI3K-Akt、Rap1、cAMP等信号通路;涉及能量代谢、免疫应激、细胞分化、神经发育、细胞骨架、信号传递等生物学过程.将获得的转录子运用Cytoscape软件构建分子相互作用网络,最终获得7个核心转录子.涉及到神经发育、次生壳形成等相关基因调控关系.
笠贝;数字化差异显示;变态相关基因;生物分子网络
0 引言
贝类的生活史中有浮游幼虫阶段,其幼虫和成体在形态结构和生活习性上差别巨大.幼虫通过变态过程转变为与成体形态结构和生活习性相同的稚贝.变态是贝类发育过程的关键环节,直接影响其种群分布、数量变动和物种进化[1].笠贝(Lottiagigantea)隶属软体动物门(Mollusca)、腹足纲(Gastropoda)、前鳃亚纲(Prosobranchia)、笠形腹足目(Patellogastropoda)、莲花青螺科(Lottiidae)(国内分类为笠贝科(Acmaeidae)),又名加利福尼亚大戚(California Limpet,Ow l Limpet),主要分布在美国加州北部到加利福尼亚半岛南部的太平洋沿岸.其壳长可达9 cm,寿命最长达20年,肉质鲜美,营养丰富,是一种重要的海洋经济软体动物.同其他贝类相比,其基因组比较小,成为研究进化和发育的重要模式物种[2].
在我国与笠贝同科的物种有,鹈足青螺(鸡爪拟帽贝,Patelloidasaccharina),主要分布在浙江以南各沿海海域;花青螺(史氏笠贝,Nipponacmeaschrenckii),主要分布于我国南、北沿海以及日本、朝鲜等地;射线青螺(细线拟帽贝,Acmaeastriata),主要分布于台湾海峡两岸、印度尼西亚等地;嫁戚(Cellanatoreuma),主要分布于山东东海沿岸到浙江北部沿岸潮间带.其中,嫁戚分布纬度与笠贝相似,且其食物链短、营养价值高、活动范围小,自然增殖年限为3龄,是理想的岩相区域性增殖贝类[3].研究笠贝的基因,构建其变态相关分子网络,对嫁戚的保护和养殖具有重要意义.
在NCBI的核酸数据库,该物种的cDNA序列众多,并且于2012年完成了基因组测序.如何利用这些海量的数据信息,成为生物科学工作者面临的一个重要问题.NCBI在线数字化差异显示工具DDD(Digital Differential Display,http://www.ncbi.nlm.nih.gov/UniGene/ddd.cgi)能够分析比较不同cDNA文库中转录子的表达差异,为充分利用大量的cDNA文库测序信息提供了一个解决途径. Blast2Go是一个综合型的序列注释分析软件,可以按照需要对DNA序列进行比对注释和分析,获得不同数据库中该序列的信息.Cytoscape是一款可图形化显示数据并具有分析和编辑功能的软件,是构建生物网络的常用软件.
本文采用DDD筛选出笠贝变态前后的差异表达的转录子,根据转录子的功能和表达量,通过Cytoscape软件构建了笠贝变态相关基因的调控网络,为合理利用丰富的网络核酸数据提供一个实例.
1 方法
1.1 文库构建
DDD基于UniGene数据库.UniGene将物种EST序列按最适标准的保守方法分配成群集(Clusters),每个群集代表唯一基因.DDD利用UniGene这个特点,比较不同cDNA文库间时间序列差异的表达谱序列,选出特定的UniGene Cluster[4].在特定状态下,cDNA文库的UniGene中存在许多差异,其中部分差异显示序列具有重要研究价值.DDD利用Fisher精确检验(The Fisher Exact Test)方法,对比发现有重要研究价值的差异显示序列.这些序列条数越多,Fisher检验值越高,检索对比得到的序列价值就越高[5].将有价值的UniGene的群集下载到本地系统,可以进行后续EST的拼接和组装工作[6].
本研究利用DDD对笠贝(L.gigantea)EST数据库进行筛选,将不同的cDNA文库按照不同发育时间分成3个文库(pool),获得在统计学上表达量有显著差异的基因的EST序列.文库构建标准及每个文库包含的ESTs数量见表1.这些差异基因的表达量数据是本文进一步数据分析的基础.
1.2 序列拼接与结果筛选
将DDD筛选的ESTs以转录子为单位打包下载,使用UniGene自身的名称“Lgi.XXX”对ESTs命名.使用信息学软件DNAstar中的Seqman功能对下载获得的所有EST序列分别进行拼接.分别将拼接获得的Contigs序列另存为fasta格式文件,并以对应的“Lgi.XXX”命名.
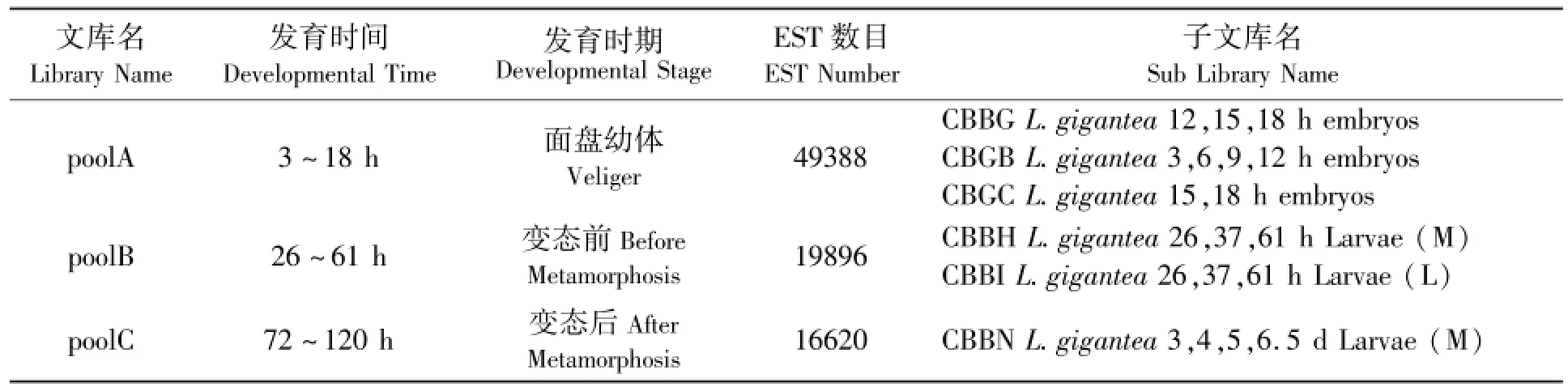
表1 DDD文库构建标准及ESTs数量Tab.1 Standards and the number of ESTs for DDD library construction
1.3 序列分析与本体论分类
Blast2Go是一个综合型的序列注释分析软件.优点是易使用、高通量,且各部分相互协作,可以自主进行调配,按照需要对数据进行整理统计并做成想要的形式,支持GO、KEGGmaps、Interpro和Enzyme Codes等数据库[7].
从Blast2Go官网(http://www.Blast2Go.com/b2ghome)下载并安装Blast2Go 2.8 PC端(500 MB),点击File中的Load FASTA File(.fasta),将所有拼接结果的fasta文件导入软件中.运行Blast中的Run BLAST,并将Blast Program设置为Blastx,Blast DB选择为nr,Blast Expectvalue设置为1.0E-3.运行Blast完毕后,再选择运行Mapping中的Run GO-Mapping,接着运行Annotation中的Run Annotation,参数设置如下:E-Value-it-filter为1.0E-6,Annotation-Cutoff为55,Go-Weihgt为5.通过点击Statistics中的各项获得相关的统计学数据.通过运行Analysis中的Make Combined Graph绘制本体论分类图,并导出其Excel格式文件.
1.4 表达趋势归类
将Blast2Go的输出结果与表达量、序列信息一一对应整合入Excel表格中.统计并制作各基因在不同时期的表达量变化趋势图.按变化趋势对基因进行分类.本文将稚贝3个变化阶段(受精到面盘幼体、面盘幼体到变态前、变态前到变态后)不同表达趋势的基因分为以下几类:
Ⅰ)表达量升高,即受精至稚贝表达量一直上升;Ⅱ)表达量降低,即受精至稚贝表达量一直下降;
Ⅲ)表达量先升高后降低,即变态阶段的表达量最高,高于变态前和变态后;Ⅳ)表达量先降低后升高,即变态阶段的表达量最低,低于变态前和变态后.
对各分类间基因变化关系进行分析,并依据William[8]的分类标准,将筛选获得的124条变态相关基因分为9大类:钙离子结合、分子相互作用、代谢、免疫应激、膜蛋白、基质蛋白、细胞骨架、核糖体组分和信号传递.结合表达趋势,获得基因功能的表达趋势分类结果.
1.5 表达量数据标准化与调控相关系数计算
对表达趋势归类中单独列出的基因的表达量数据进行对数转换后做标准化处理[9].公式为:
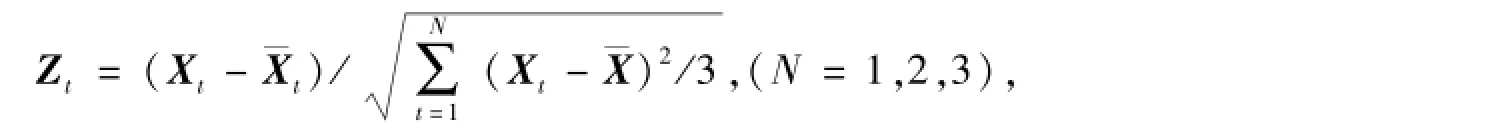
其中:Xt为该基因t时序时对数处理后的表达量向量,t为表达时序,N为t的具体取值范围,Zt为标准化的表达量向量.利用Pearson相关系数评估不同基因之间表达量相似性,以进一步确定它们之间是否有调控关系,筛选出起调控作用的基因[10].
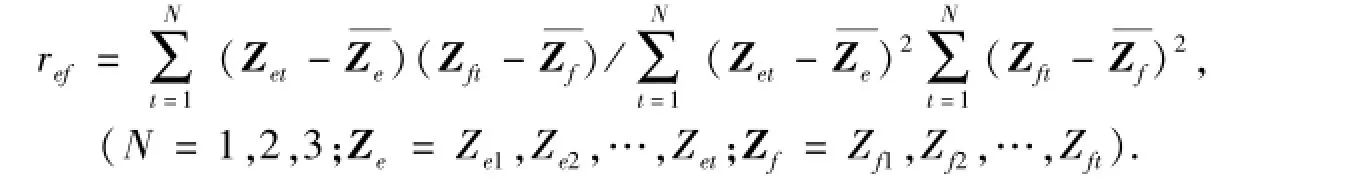
使用矩阵中任意基因E和F的标准化表达量向量计算E、F的相关系数ref.公式为:当r值为1时,说明两条基因是正调控;当r值为-1时,说明两条基因是负调控;当r值为0时,说明两条基因没有调控关系.r值越接近1或-1,两条基因相关性越显著[9].使用SPASS软件计算r值,并检验其显著性P,取P<0.05的r值(显著差异)作为网络构建的依据[9,11-12].
1.6 核心调控网络生成
将获得的基因、Blast2Go结果、表达量数据、调控关系和相关系数运用CSV文件编辑器软件,按照Cytoscape格式(基因1、基因间的关系、基因2)编辑生成genelist格式的文件,将文件导入Cytoscape中生成初步调控网络图[13].从初步网络中筛选出与神经发育、细胞分化、次生壳形成、能量代谢相关的基因,构建核心调控网络.
2 结果
2.1 序列分析与分类
用Blast2Go软件对序列拼接和筛选得到的203条序列进行Blast、Mapping和Annotation分析,剔除无结果序列(24条)、结果重复及错误序列(13条)和只有假定蛋白序列(42条),最终选出具有有效功能注释的序列124条.通过基因本体论(GO)分类,可以将这些基因归为细胞组分(cellular component)、分子功能(molecular function)和生物学过程(biological process)3类,分别占到总基因(203条)的66.00%、55.66%和88.18%(如图1所示).
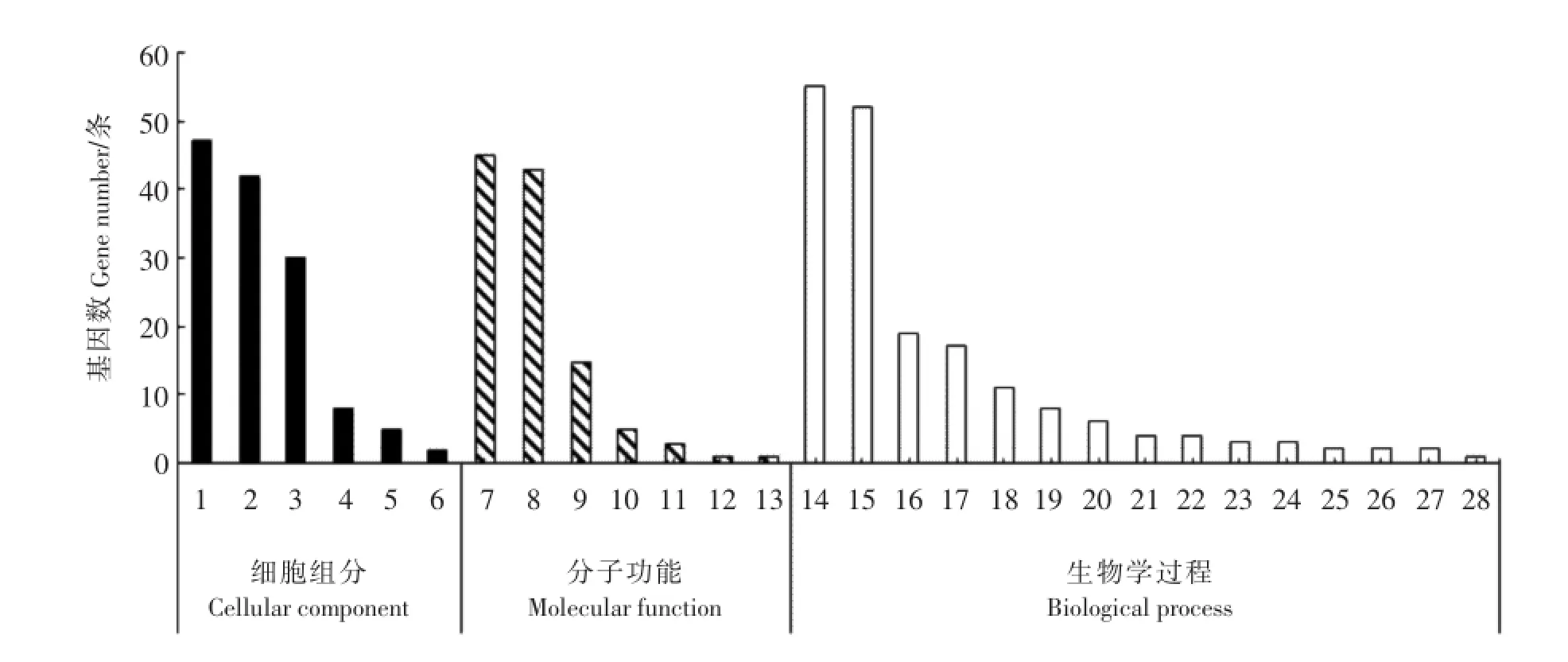
图1基因GO注释一级和二级分布Fig.1 Distribution ofGO annotations of identified genes tha tbe long to bo th the prim ary and secondary func tions
在134条具有细胞组分注释的基因中,细胞(cell)、细胞器(organell)和高分子配合物(marcromolecular complex)条数最多,分别为47条、42条和30条,占差异基因的23.15%、20.69%和14.78%.有分子功能注释的基因有113条,其中具有结合(binding)、催化活性(catalytic activity)和结构分子活性(structuralmolecule activity)功能的基因条数最多,分别为45条、43条和15条,占差异基因的22.17%、21.18%和7.39%.具有生物学过程注释的基因有179条,其中细胞过程(cellular process)、代谢过程(metabolic process)和单一的生物过程(single-organism process)条数最多,分别为55条、52条和19条,占差异基因的30.73%、29.05%和10.61%.
2.2 表达趋势归类及网络构建
根据表达趋势划分的4类基因(见表2)中,Ⅰ类(表达量升高)基因有42条,占总数的33.87%,其中代谢最多(12条),信号传递次之(8条),基质蛋白和免疫应激并列第三(5条),核糖体组分最少(1条).Ⅱ类(表达量降低)基因有50条,占总数的40.32%,其中,代谢最多(17条),分子相互作用基因次之(10条),细胞骨架基因第三(8条),核糖体组分基因最少(1条).Ⅲ类(变态表达量最高)基因有28条,其中核糖体组分基因最多,有7条;信号传递基因次之,有5条;没有发现钙离子结合功能的基因.IV类(变态表达量最少)基因仅有4条,其中3条属于代谢基因,1条属于信号传递基因.
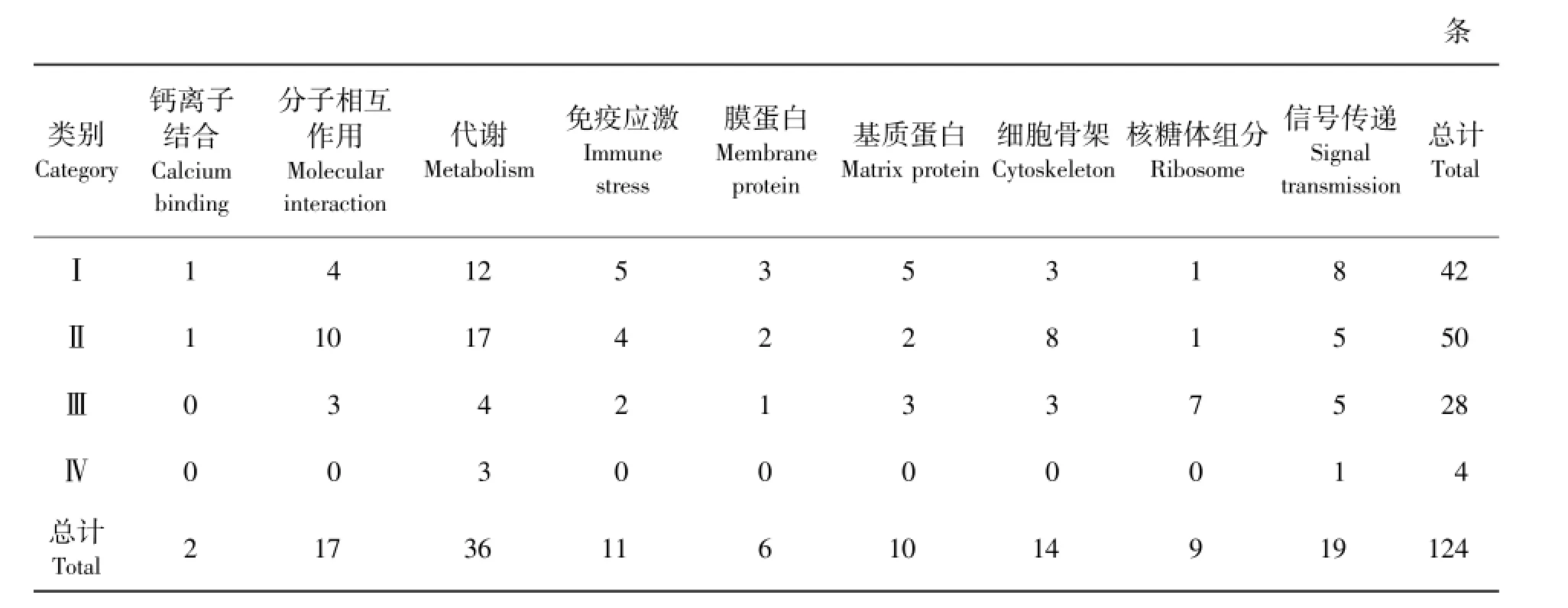
表2 基因表达量及功能分类Tab.2 Expression and functiona l classification of identified genes
2.3 分子相互作用网络构建
用统计学软件SPSS对所有基因两两之间的表达量相关系数r进行计算.结果共获得7099个配对,其中显著性结果有560个,占总结果数的7.9%;极显著结果有216个,占总结果数的3.0%.运用Excel提取P<0.05的r值,并使对应的基因一一配对.将前期获得的注释结果、变化趋势和相关系数经CSV文件编辑器处理后,提交到Cytoscape软件中生成初步相互作用网络.从初步网络中手工筛选神经发育、组织分化、次生壳形成、能量代谢相关基因38个,再以这38个基因构建核心调控网络(见表3、图2).
由表3、图2可见,网络有7个核心基因,分别是辅酶合成加氧酶7同系物(coq7)、刺猬蛋白(hh)、下连合器脊椎蛋白(sspo)、DNA结合蛋白抑制剂(id2)、G蛋白亚基1(gng1)、神经增殖及分化和控制蛋白1(npdc1)、未知软体动物壳蛋白13(lusp-13).以它们为中心,分别形成了2个大集团(集团1和集团2)和1个小集团(集团3).集团1主要由III类和IV类基因组成.核心分子coq7为III类基因,与其他相互作用分子有正相关关系,并且这些相互作用分子几乎都为Ⅲ类基因.另一个核心分子hh与集团2的核心分子npdc1具有正相关关系,将两个集团联系起来.集团2主要由I类和II类基因组成,形成了2条具有正相关关系的相互作用链dpys-flot1-try-3-csad-npdc1-sspo和cyt-cdynll2-nxnl-id2-h3f3a-npdc1,1条具有负相关关系的作用链dpys-gabarap-nlp-dynlt1-if-h3f3a-npdc1.此外,gng1等3个变态期表达量最高的基因与钙调蛋白(cam)形成一个具有正相关作用的小集团.

表3 核心网络中基因注释结果Tab.3 Annotation of genes of core network
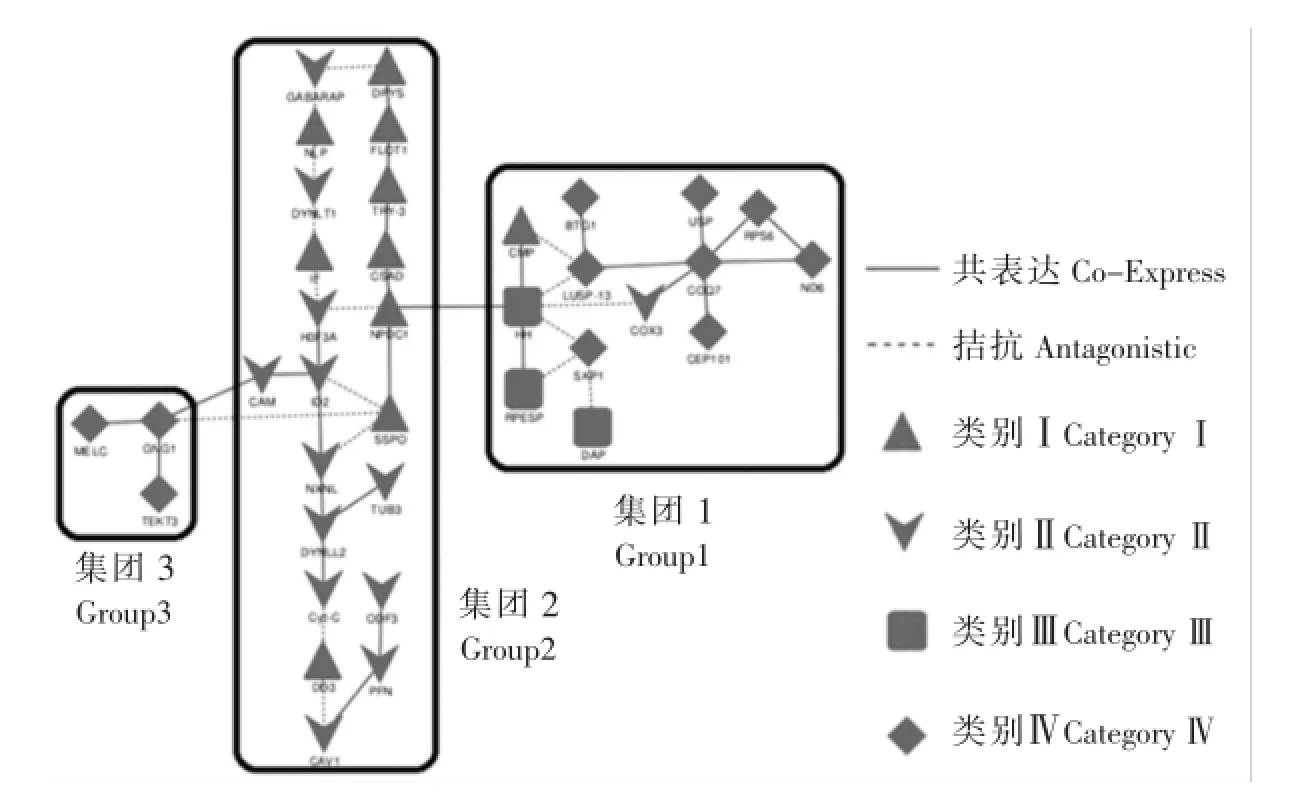
图2笠贝幼虫变态相关基因相互作用核心网络Fig.2 Core network of L.gigantea larva lofmetamorphosis-related genes
3 讨论
利用表达量数据构建分子调控网络是分子生物学系统研究分子相互作用的新方法,它能有效利用几何级数增长的生物信息数据,从更宏观、更全面的角度去研究各类生物学问题[14].NCBI的DDD基因差异表达分析,是通过比较不同cDNA文库中同一基因EST数量,来获得这一基因的表达差异的,其本质上与比较二代测序转录组差异基因分析是相同的.而且DDD利用的cDNA文库是NCBI整理认可的,文库测序数据皆由实验获得,其可信度有保障[15].目前有很多利用二代测序转录组的方式进行基因变态差异的研究,定量PCR的结果与测序分析具有很好的一致性[16-18].这表明基于测序的基因差异表达分析具有较高的可信度.本文的分析方法是对爆炸性增长的测序数据发掘利用的一次尝试.
笠贝与鲍同属于腹足类,变态过程类似.笠贝已经完成基因组测序,转录组数据非常丰富.利用笠贝丰富的核酸序列资源,能够方便地获得笠贝幼虫变态的基因表达过程.这对分析相近物种鲍的变态分子机制,具有重要参考意义.笠贝是目前在DDD中与杂色鲍唯一近缘的物种,其原产地在北美,材料获取困难,因此本研究结果并未进行定量PCR的验证.但是笔者根据本文预测结果,在杂色鲍中进行相关基因的定量PCR检测,其结果与构建的网络一致性很好.这表明DDD的分析结果具有较高的可靠性.利用DDD中的近缘物种进行分析,能够提高研究的目的性.
在所获得差异表达基因中,参与细胞学过程的基因最多,细胞、细胞器相关基因占总体比例最高,表明幼虫变态过程中细胞分化增殖剧烈,这与变态过程中新组织的形成和幼虫特有组织的崩解凋亡相关.参与代谢过程的基因数量次之,表明变态过程中能量分配对变态成功率有重要影响,这与Moran和Shilling的研究结果相符[19-20].贝类幼虫变态前能量的储备是变态成功的关键,一般个体较大的幼虫变态率较高[21].处于变态期的幼虫同初期面盘幼虫相比,自身体重增加显著,其为保持浮游状态需要消耗更多能量.变态期幼虫运动活跃,面盘纤毛摆动力度和频率都明显增加.这些因素可能是代谢相关基因占总体比重较高的部分原因.
根据表达趋势划分的4类基因中,I类基因可能与幼虫的发育成熟关系较大,它们为变态后幼虫的存活进行物质和功能上的储备.如,ODC是多胺产生的关键酶.多胺参与昆虫卵黄发生、胚胎发育、变态、行为等生理调节过程[22];CSAD是牛磺酸和亚牛磺酸合成的关键酶,二者能促进脂肪及脂溶性维生素的消化吸收.CYP356A1可能与外源性生物转化、类固醇代谢有关[23].这些基因表达量的上升表明幼虫在消化系统方面一直在完善,与鲍幼虫具有“‘Anticipatory’Developmental Program”的现象一致[24].II类基因与胚胎发育关系密切,所以随着发育进程结束表达量逐渐降低.Ⅱ类基因中代谢类的基因最多,其中变化最剧烈的5个基因cox2、cox3、cyt-c、nd2、nd3都参于氧化磷酸化过程,17条代谢类基因中有12条与氧化磷酸化有直接关系.这表明笠贝胚胎发育至面盘幼虫阶段能量消耗巨大,且与Vavra等[25]在红鲍中的研究结果一致.III类基因在变态过程表达量最高,可能是变态过程的重要参与者.Ⅲ类基因中核糖体组分最多,Dong等[26]在研究棉铃虫(Helicoverpa armigera)的变态过程时也发现核糖体组分基因差异的表达,可能如文献[21]所说是与作为细胞增殖和细胞凋亡的调节蛋白直接参与了变态过程有关;信号传递基因数量次之,表明这一时期幼虫的信号传递系统进一步完善,能够响应外界变态诱导因子启动变态过程,这是贝类变态的特点之一[21,28]. IV类基因只有4个,分别是dap、hh、abat、rpesp,它们可能是变态过程的抑制因子.例如dap与TGFB相关,共同调节细胞凋亡[28];hh能影响MAPK通路,抑制脂肪细胞发育[29].在核心分子网络中,集团1主要由III类和IV类基因组成,coq7和hh是其核心分子.与coq7相关的基因主要以能量代谢为主,如usp是蜕皮激素受体靶标基因,参与脂肪代谢,而nd6和cox3参与氧化磷酸化过程;与hh相关的基因有cmp、lusp-13、rpesp、skp1、dap等,它们与细胞周期有密切联系.集团2以npdc1为核心分子,形成了2条具有正相关关系的相互作用链和1条具有负相关关系的作用链,多与神经发育有关.
目前关于软体动物的各类研究成果,相对于人、鼠等高等生物来说比较少,数据的可获得性和数据质量在一定程度上限制了网络的预测精度.另外,本文中选取的神经发育、组织分化、次生壳形成、能量代谢相关基因的限制范围较小,对最终的结果有一定影响,这也是本研究的局限之一.随着生物信息学技术的不断进步和可利用的生物信息学研究方法的完善,将不断有更简便、高效、准确的方法诞生,同时数据库的样本丰度也会不断提高.下一步,笔者将会使用更先进的数据筛选和关联模型,通过增加差异化表达数据来源,参考同源比对、亚细胞定位、Gene Ontology信息来进一步提高网络模型的预测精度.
[1]UNDERWOOD A,KEOUGH M J.Supply-side ecology:the nature and consequences of variations in recruitmentof intertidal organisms[J].Marine Community Ecology,2001,87(1):183-200.
[2]VEENSTRA JA.Neurohormones and neuropeptides encoded by the genome ofLottiagigantea,with reference to other mollusks and insects[J].General and Comparative Endocrinology,2010,167(1):86-103.
[3]王志铮,吴常文.浙北沿海嫁戚Cellanator年龄与生长的研究[J].浙江海洋学院学报:自然科学版,2000(4):316-323.
[4]SCHEURLE D,DEYOUNGM P,BINNINGER D M,et al.Cancer gene discovery using digital differential display[J]. Cancer Research,2000,60(15):4037-4043.
[5]OLESEN C,HANSEN C,BENDSEN E,et al.Identification of human candidate genes formale infertility by digital differential display[J].Molecular Human Reproduction,2001,7(1):11-20.
[6]顾朝辉.生物信息挖掘新基因预测平台的建立与TSEG-3的克隆和功能初步研究[D].武汉:华中科技大学,2010.
[7]CONESAA,GÖTZS,GARCÍA-GÓMEZ JM,etal.Blast2go:a universal tool for annotation,visualization and analysis in functional genomics research[J].Bioinformatics,2005,21(18):3674-3676.
[8]WILLIAMSEA,DEGNAN B M,GUNTER H,et al.Widespread transcriptional changes pre-empt the ritical pelagicbenthic transition in the vetigastropodHaliotisasinina[J].Molecular Ecology,2009,18(5):1006-1025.
[9]栾德琴.鸡肌肉生长相关基因的表达与肌苷酸关键酶基因网络调控的构建[D].扬州:扬州大学,2012.
[10]刘天飞,唐国庆,李学伟.不同实验类型的基因表达数据聚类分析方法研究[J].畜牧兽医学报,2009,40(2):180-184.
[11]崔焕先.肉鸡肌内脂肪形成的分子调控网络及相关基因研究[D].北京:中国农业科学院,2011.
[12]张永新,李子健,闫洁,等.利用苯肾上腺素诱导新生大鼠心肌细胞肥大基因表达谱构建其调控网络[J].中国病理生理杂志,2009(11):2081-2087.
[13]NEEDHAM C J,MANFIELD IW,BULPITT A J,et al.From gene expression to gene regulatory networks inArabidopsishaliana[J].Bmc Systems Biology,2009,3(1):85.
[14]张哲,荣起国.数据整合方法构建大鼠分子调控网络[J].系统仿真学报,2009(5):1479-1483.
[15]HE T,CHEN J,ZHANG J,etal.Sarp19 and Vdg3 gene familiesare functionally related duringabalonemetamorphosis[J].Dev Genes Evol,2014,224(4):197-207.
[16]ZHANG X,MAO Y,HUANG Z,et al.Transcriptome analysis of the octopus vulgaris central nervous system[J]. Plos One,2012,7(6):825-833.
[17]QIN J,HUANG Z,CHEN J,et al.Sequencing and denovo analysis ofCrassostreaangulata(Fujian oyster)from 8 different developing phases using454 gsflx[J].Plos One,2012,7(8):1322-1337.
[18]HUANG Z,CHEN Z,KE C,et al.Pyrosequencing ofHaliotisdiversicolortranscriptomes:insights into early developmentalmolluscan gene expression[J].Plos One,2012,7(12):1599-1614.
[19]SHILLING FM,HOEGH-GULDBERG O,MANAHAN D T.Sources of energy for increased metabolic demand during metamorphosis of the abaloneHaliotisrufescens(mollusca)[J].The Biological Bulletin,1996,191(3):402-412.
[20]MORAN A L,MANAHAN D T.Energy metabolism during larval development of green and white abalone,HaliotisfulgensandH.sorenseni[J].The Biological Bulletin,2003,204(3):270-277.
[21]王国栋,张丽莉,王艺磊.鲍幼虫变态分子机制的研究进展[J].集美大学学报:自然科学版,2012,17(2):101-108.
[22]SPARKSR B,VASKE D,LILLEBERG S,et al.Temporal expression of ornithine decarboxylase in developing embryos ofMuscadomestica[J].Archives of Insect Biochemistry and Physiology,1991,16(3):177-187.
[23]TOLEDO-SILVA G D,SIEBERTM N,MEDEIROS ID,et al.Cloning a new cytochrome P450 isoform(cyp356a1)from oysterCrassostreagigas[J].Marine Environmental Research,2008,66(1):15-18.
[24]DEGNAN BM,MORSE D E.Developmental and morphogenetic gene regulation inHaliotisrufescenslarvae atmetamorphosis[J].American Zoologist,1995,35(4):391-398.
[25]VAVRA J,MANAHAN D T.Protein metabolism in lecithotrophic larvae(gastropoda:Haliotisrufescens)[J].The Biological Bulletin,1999,196(2):177-186.
[26]DONGD,HEH,CHAIL,et al.Identification of genes differentially expressed during larvalmolting andmetamorphosis ofHelicoverpaarmigera[J].Bmc Developmental Biology,2007,7(1):73.
[27]柯才焕,冯丹青.海洋底栖动物浮游幼体附着和变态的研究[J].厦门大学学报:自然科学版,2006,45(A02):77-82.
[28]李仲诚,阮礼波,杨帆,等.死亡相关凋亡诱导蛋白激酶2(DRAK2)研究进展[J].生命科学,2014,26(8):823-828.
[29]CHEN J,TANG Q.Hedgehog signaling pathway and adipocyte development[J].Chinese Journal of Biochemistry and Molecular Biology,2011,27(1):6-10.
(责任编辑 朱雪莲 英文审校 张子平)
Indentification of M etamorphosis Related Genes and
Development of M olecular Networks ofLottiagiganteaLarval
YIN Cheng1,2,ZHANG Li-li1,2,WANG Guo-dong1,2,WANG Yi-lei1,2
(1.Fisheries College,Jimei University,Xiamen 361021,China;2.Key Laboratory of Healthy Mariculture for the East China Sea,Ministry of Agriculture,Xiamen 361021,China)
By using the NCBIonline Digital Differential Display tool,203 differentially expressed genes in metamorphosis ofLottiagigantealarvae were identified from ESTs database ofL.gigantea.About 124 genes had Blast2Go annotationswhich were classified into cellular components,molecular functions and biological processes by GO(Gene Ontology).These genes are members of some signal pathways,such as MAPK,Ras,PI3K-Akt,Rap1,cAMP,et al,and refer tomany biological processes,such as energymetabolism,immunological stress,cell differentiation,neurodevelopment,cytoskeleton,signal transmission. These 124 geneswere used to buildmolecular interaction network by Cytoscape software.A core network,including seven core genes,was showed.The network involved in nerve development,tissue differentiation,secondary shell formation and energymetabolism,and revealed a possible molecular mechanism ofmetamorphosis.
Lottiagigantea;Digital Differential Display;metamorphosis related genes;molecular interaction network
Q 959.212+.2;Q 789
A
1007-7405(2015)05-0339-09
2015-02-16
2015-04-15
国家自然科学基金项目(41006105,41176152)
尹诚(1988—),男,硕士生,从事贝类功能基因研究.通信作者:王国栋(1977—),男,副教授,博士,从事贝类发育遗传方向研究.E-mial:gdongwang@163.com.
