胡黄连苷Ⅱ对脑缺血损伤后神经细胞凋亡和超微结构的影响
2015-06-09王婷婷李晓丹张美增郭云良
王婷婷,赵 丽,李晓丹,张美增,郭云良
(青岛大学附属医院脑血管病研究所,山东 青岛 266003)
胡黄连苷Ⅱ对脑缺血损伤后神经细胞凋亡和超微结构的影响
王婷婷,赵 丽,李晓丹,张美增,郭云良
(青岛大学附属医院脑血管病研究所,山东 青岛 266003)
目的 探讨胡黄连苷Ⅱ对脑缺血损伤后神经细胞凋亡和超微结构的影响。方法 成年健康♂ Wistar大鼠60只,应用线栓法建立大鼠脑缺血模型,经腹腔注射胡黄连苷Ⅱ (20 mg·kg-1) 干预治疗,改良神经功能评分(mNSS)评价动物神经行为功能,氯化三苯基四氮唑染色观察脑梗死体积,HE染色和透射电镜观察脑组织病理和超微结构,原位末端标记法检测细胞凋亡,免疫组织化学和Western blot检测p-ERK1/2表达水平。结果 大鼠脑缺血损伤后表现出神经功能障碍和脑梗死病灶,皮质区神经元和血脑屏障结构损伤较重,皮质区凋亡细胞数量和p-ERK1/2蛋白表达较对照组明显增多(P<0.05)。治疗组大鼠mNSS评分和脑梗死体积较模型组明显降低(P<0.05),皮质区神经元和血脑屏障损伤减轻,凋亡细胞与p-ERK1/2表达水平较模型组明显降低(P<0.05)。结论 胡黄连苷Ⅱ可能抑制神经细胞凋亡,改善缺血区脑组织的形态结构,促进大鼠神经行为功能恢复。
胡黄连苷Ⅱ;脑缺血;凋亡;超微结构;大鼠; p-ERK1/2
脑缺血损伤引发缺血半暗带区神经细胞凋亡[1]。早期研究证实,脑缺血损伤后脑组织处于低氧环境中,神经细胞损伤表现为坏死或凋亡,细胞外信号调节蛋白激酶1/2(ERK1/2)通路的激活能够促进细胞的增殖、分化与修复,减轻神经细胞损伤[2-3]。但后来实验证明,ERK1/2通路在脑缺血损伤中能够促进炎症反应、介导神经细胞凋亡[4]。ERK1/2通路抑制剂和钙通道阻滞剂氯胺酮可抑制脑缺血大鼠ERK1/2通路的激活,下调miRNA-21和MMP-9表达,降低炎症反应,保护血脑屏障损伤,减轻脑水肿[5]。本课题组研究证明,胡黄连苷Ⅱ在脑缺血损伤的多个环节中发挥作用,具有抗氧化、抗炎、减轻脑水肿、抑制神经细胞凋亡的作用[6-10],但细胞的形态结构依然是评价神经系统损伤和修复程度的可靠指标。因此,本实验研究胡黄连苷Ⅱ对脑缺血损伤后神经细胞凋亡及其超微结构的影响,进一步验证黄连苷Ⅱ对脑缺血损伤的保护作用。
1 材料与方法
1.1 动物模型成年健康♂ Wistar大鼠60只,体质量220~250 g,SPF级,青岛市药品检验所实验动物中心提供[SCXK(鲁)20100010]。实验前将动物置于实验室适应环境,饲养7 d,自由进食、饮水,室温(25±2)℃,自然光照。随机选择15只大鼠作为对照组,其余45只大鼠运用线栓法[11]经左侧颈外-颈内动脉插线建立大脑中动脉阻塞(MCAO)模型。术前12 h禁食,腹腔注射10%水合氯醛(3 mL·kg-1)麻醉动物,仰卧位固定,无菌操作。大鼠术后2 h应用改良神经功能评分(mNSS)[12]法评分,将评分为7~12分的大鼠视为成功模型纳入实验,不符合标准和术后2 h死亡的15只动物剔除。将成功的30只动物模型随机分为模型组和治疗组,每组15只,对照组动物除不插线进入颅内外,余操作同实验组。
1.2 干预措施用生理盐水将胡黄连苷Ⅱ(CAS No:39012-20-9,纯度>98%,分子量:512,天津奎青有限公司)配制成10 g·L-1的溶液,治疗组大鼠在缺血后2 h腹腔注射胡黄连苷Ⅱ(20 mg·kg-1)[13-14],模型组和对照组同步腹腔注射等体积的生理盐水。
1.3 评价指标
1.3.1 行为测试 治疗前和治疗后24 h,应用mNSS法评定动物的神经行为功能,0~18分,评分越高说明动物神经行为功能损伤越重,反之则较轻微。
1.3.2 TTC染色 治疗后24 h,每组随机选取5只动物,经心脏灌注生理盐水200 mL取脑,利用大鼠切脑模具由前向后连续冠状切片,每片厚2 mm,共计5片,置20 g·L-1的氯化三苯基四氮唑(TTC)磷酸盐缓冲液中浸泡 10 min,37℃避光,正常脑组织呈均匀红色,梗死组织呈白色,拍照。应用Adobe PhotoShop CS测量脑梗死体积,脑梗死体积(cerebral infract volume,CIV)=经过视交叉平面的脑梗死面积/该层同侧半球面积。
1.3.3 HE染色 治疗后24 h,每组随机选取5只动物,经心脏灌注4%多聚甲醛溶液200 mL取脑,切取缺血部位脑组织置4%多聚甲醛溶液后固定2 h,蒸馏水浸泡4 h。常规脱水、透明、包埋,连续冠状位切片,厚度为5 μm,贴于防脱载玻片上,4℃保存。石蜡切片常规脱蜡水化,苏木精染色5 min,1%盐酸酒精分色20 s,l%氨水返蓝30 s,伊红染色5 min;梯度乙醇脱水,二甲苯透明,中性树胶封片。显微镜(Leica DMI4000B,德国)下观察,神经细胞核呈蓝色,细胞质呈不同程度的红色。在400倍显微镜下,每张切片随机观察皮质区4个不重叠的视野计数细胞,取其均值。以变性细胞指数(denatured cell index,DCI)=变性细胞数/细胞总数表示损伤程度。
1.3.4 电镜观察 从上述动物缺血皮质区取少量脑组织,切成1 mm×1 mm×1 mm小块,置于2.5%戊二醛中固定24 h,0.1 mol·L-1磷酸缓冲液(PBS)漂洗10 min×3次;锇酸后固定1 h,PBS漂洗10 min×3次,4℃冰箱过夜;梯度丙酮脱水,环氧树脂Epon812包埋。修块后应用超薄切片机(Leica EM UC6,德国)切成50 nm超薄切片,置于聚乙烯醇缩甲醛膜(Formvar膜)载网(铜网)上,4℃保存。预先配制3%醋酸铀饱和酒精溶液和6%枸橼酸铅染液。将1滴3%醋酸铀饱和酒精溶液(pH=3.5)滴在放有洁净蜡片的培养皿中,然后将带有切片的载网覆盖在染液上,使有切片的一面与染液接触,染色30 min,双蒸水洗涤切片10 min×3次,吸干水分,用同样方法6%枸橼酸铅染液(pH=12)染色5 min,去二氧化碳双蒸水冲洗10 min×3次,吸干水分,室温干燥,透射电镜(JEM-1200EX,日本)观察神经元的超微结构。
1.3.5 TUNEL染色 石蜡切片常规脱蜡水化,按TUNEL细胞凋亡检测试剂盒(KGA7025-50assays,凯基生物公司)说明书预处理,促渗、封闭、通透、制备阳性片、POD标记、DAB显色和苏木精复染等。光镜下细胞核有棕色颗粒者为凋亡细胞。阴性对照样本不加TDT酶,不出现阳性着色。在400倍显微镜下,每张切片随机观察皮质区4个不重叠视野,计数细胞,取其均值。以凋亡细胞指数(apoptotic cell index,ACI)=凋亡细胞数/细胞总数,表示细胞凋亡程度。
1.3.6 免疫组化 p-ERK1/2兔抗鼠单克隆一抗(1 ∶100,#4370)由Cell Signaling Technology公司提供,二步法免疫组化检测试剂盒(PV-6001)由北京中杉金桥生物公司提供。石蜡切片常规脱蜡水化,阻断内源性过氧化物酶、柠檬酸盐缓冲液抗原修复、37℃一抗孵育60 min、室温二抗孵育60 min、DAB显色、苏木精复染、中性树胶封片。光镜下阳性细胞呈棕黄色。阴性对照切片以0.1 mol·L-1的PBS替代一抗孵育,无阳性细胞出现。在400倍显微镜下,每张切片随机观察皮质区4个不重叠视野,细胞计数,取其均值。以阳性细胞指数(positive cell index,PCI)=阳性细胞数/细胞总数,表示p-ERK1/2的表达强度。
1.3.7 Western blot 治疗后24 h,每组取5只动物,切取缺血部位脑组织100 mg,按脑组织和细胞裂解液10 mg ∶100 μL的比例加入细胞裂解液(1 mL裂解液+10 μL PMSF,No. P0013B,碧云天生物技术研究所)1 mL,4℃冰浴中研磨,置于4℃摇床0.5~1 h充分裂解,4℃离心机(Eppendorf 5801型,德国)12 000 r·min-1离心15 min,去沉淀组织留上清液,BCA法测定蛋白浓度,最后加入SDS-PAGE Sample Loading Buffer(5×)充分混匀,置95~97℃水浴中加热5 min,-80℃保存。取蛋白样品20 μg,SDS-PAGE凝胶电泳分离蛋白,半干法转膜(Bio-Rad公司)。将PVDF膜分别浸入含质量分数为10%的脱脂奶粉的TBST封闭液,室温封闭1 h,用质量分数5%BSA的TBST配制兔抗鼠p-ERK1/2单克隆抗体(1 ∶2 000)一抗稀释液,4℃摇床孵育过夜,TBST洗膜10 min×3次,用质量分数为5%的脱脂奶粉的TBST配制HRP标记的羊抗兔二抗稀释液(1 ∶10 000, Ab136817,Abcam公司)室温孵育1 h,TBST洗膜10 min×3次。最后加入200 μL显影工作液混匀,化学发光多色荧光及活体成像系统(Vilber Fusion FX7,法国)曝光显影,Quantity one软件分析各条带灰度值。将PVDF膜浸入Restore Western Blot Stripping Buffer(21062,Thermo公司)中10min,洗去一抗二抗,再重复封闭、质量分数为5%BSA的兔抗鼠ERK1/2多克隆抗体(1 ∶1000, #9102,Cell Signal Technology, USA)TBST一抗稀释液4℃摇床孵育过夜、二抗室温孵育1 h,发光显影,测定各条带灰度值等步骤。计算目的蛋白相对值(relative value of protein,RVP)=p-ERK1/2灰度值/ERK1/2灰度值。重复测定5次,取其均数。
1.4 统计学分析应用SPSS 17.0统计软件进行数据统计分析。多组间比较采用单因素方差分析及最小显著差数法(LSD)两两比较。
2 结果
2.1 神经行为功能缺损评分对照组大鼠活动正常,造模后动物表现不同程度的神经行为功能障碍。治疗前后自身比较,治疗组大鼠mNSS评分明显下降(t=5.17,P<0.05)。治疗后治疗组大鼠mNSS评分较模型组明显降低(t=6.73,P<0.05)。见Tab 1。
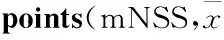
Tab 1 Modified neurological severity score ±s,n=15)
*P<0.05vscontrol group;#P<0.05vsmodel group;△P<0.05, self-compared between before and after treatment
2.2 脑梗死体积TTC染色显示,对照组大鼠脑组织呈均匀红色,未见明显梗死灶。模型组大鼠脑组织可见大片白色梗死病灶(79.82±7.56)。治疗组大鼠脑梗死体积(65.32±6.90)较模型组明显缩小(t=3.88,P<0.05)。见Fig 1。
2.3 组织病理HE染色显示,对照组大鼠皮质区神经细胞胞体较大,胞核呈嗜碱性蓝色,胞质呈嗜酸性紫红色,排列整齐,结构完整,着色均匀。模型组大鼠皮质区神经细胞排列紊乱,胞核固缩、着色加深,变性坏死后形成大量空泡,DCI(0.75±0.11)较对照组(0.10±0.07)明显升高(t=11.71,P<0.05)。治疗组大鼠皮质区神经细胞损伤较模型组减轻,DCI(0.37±0.11)较模型组明显降低(t=6.91,P<0.05)。见Fig 2。
2.4 超微结构对照组大鼠皮质神经元轮廓清晰,核大而圆,染色质均匀,胞质清晰(Fig 3A1),高倍镜下双层核膜清晰完整,胞质内线粒体内外膜完整、内膜凹陷形成嵴,高尔基体扁平囊周围有数个大小不等的囊泡,溶酶体、内质网等细胞器散在分布、结构完整(Fig 3A2)。血管内皮细胞光滑平整,内皮细胞、基底膜以及胶质细胞足突细胞接触紧密(Fig 4A1);高倍镜下内皮细胞、基底膜以及胶质细胞足突细胞层次分明,连接紧密(Fig 4A2)。模型组大鼠皮质神经元形状不规则,细胞核内染色质凝聚固缩,细胞质溶解,周围有空泡形成(Fig 3B1),高倍镜下细胞核内染色质凝聚,核膜溶解,胞质内细胞器溶解消失(Fig 3B2)。血管周围胶质细胞溶解消失,管腔凹陷、内壁不平滑(Fig 4B1),高倍镜下内皮细胞肿胀,基底膜增厚,层次不清(Fig 4B2)。治疗组大鼠神经元损伤较模型组有所减轻(Fig 3C1~C2)。血管内皮细胞、基底膜平滑完整、层次清晰(Fig 4C1~C2)。

Fig 1 Cerebral infarct volume in each group by TTC staining (n=5)
A: Control group; B: Model group; C: Treatment group.*P<0.05vscontrol group;#P<0.05vsmodel group

Fig 2 Morphology and structure of nerve cells in cortex in different groups(HE×400)
A: Control group; B: Model group; C: Treatment group. →indicated denatured cells.*P<0.05vscontrol group;#P<0.05vsmodel group
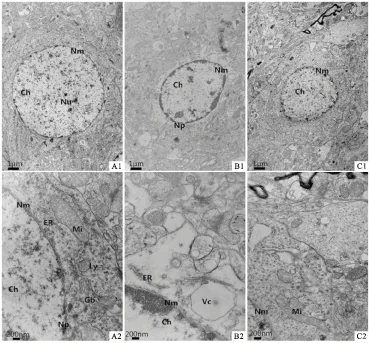
Fig 3 Ultra-structure of neuron in cortex, EM×5k, 20k
A: Control group; B: Model group; C: Treatment group; Nu: Nucleolus; Np: Nuclear pore; Nm: Nuclear membrane; Ch: Chromatin; Mi: Mitochondria; ER: Endoplasmic reticulum; Gb: Golgi complex; Ly: Lysosome; Vc: Vacuole
2.5 细胞凋亡TUNEL检测显示,对照组大鼠皮质区仅有少量凋亡细胞(ACI=0.11±0.07);模型组大鼠皮质区凋亡细胞(ACI=0.59±0.10)较对照组明显增多(t=9.16,P<0.05),排列紊乱,胞体固缩,周围空隙增大;治疗组大鼠皮质区凋亡细胞(ACI=0.33±0.07)较模型组明显降低(t=5.03,P<0.05)。见Fig 5。
2.6 免疫组化对照组大鼠神经细胞p-ERK1/2蛋白表达较弱(PCI=0.08±0.03),着色较浅。模型组大鼠p-ERK1/2表达(PCI=0.54±0.11)明显高于对照组(t=8.47,P<0.05)。治疗组大鼠p-ERK1/2蛋白表达(PCI=0.34±0.10)明显低于模型组(t=3.74,P<0.05)。见Fig 6。
2.7 Western blotWestern blot检测显示,MCAO造模后模型组大鼠皮质神经元p-ERK1/2蛋白表达(RVP=0.60±0.10)升高,明显高于其他两组(t=6.641、3.553,P<0.05)。治疗组p-ERK1/2表达(RVP=0.38±0.08)较模型组有所降低,但仍高于对照组(RVP=0.19±0.11,t=3.087,P<0.05)。见Fig 7。
3 讨论
急性缺血性脑中风发作的几分钟之内,脑缺血区血流量急剧减少,神经细胞受损,表现为坏死或凋亡,各种生长因子、氧化应激、细胞内Ca2+水平等信号激活,介导细胞增殖、分化、凋亡等生理过程[2]。实验证实[15],ERK1/2通路可以介导细胞凋亡,具体机制主要分为细胞内途径和细胞外途径[1]。细胞内途径主要是某些特定的刺激引起的,其可导致线粒体细胞色素C的释放以及caspase-9等的激活[16]。细胞外途径主要是通过激活TNF-α、Fas、caspase-8等的受体来激活细胞内途径[17]。Namura等[18]实验证实,ERK1/2通路抑制剂U0126能够降低前脑缺血或者局灶性缺血模型沙鼠的脑梗死面积,保护神经系统,并且对缺氧环境中的原代皮质神经元细胞也表现出一定的保护作用。此外,有报道证实,ERK1/2的激活可以直接导致神经细胞的肿胀、坏死,而该过程独立于caspase家族的激活[19]。
本课题组前期研究显示,在脑缺血损伤中,中药胡黄连的主要活性成分胡黄连苷Ⅱ能够清除自由基、抗氧化;通过下调MMP-9、AQP4蛋白的表达、抑制TLR4-NFκB信号传导通路,缓解脑水肿、改善大脑的神经功能[20];下调caspase-3和PARP表达,抑制脑缺血/再灌注损伤诱导的细胞凋亡[6-10];其治疗脑缺血的最佳时间和剂量为缺血后1.5~2 h腹腔注射10~20 mg·kg-1[21-22]。本实验结果表明,脑缺血损伤后,皮质缺血区神经元ERK1/2激活而诱导细胞凋亡,脑梗死体积增大,神经元和血脑屏障结构损伤,动物出现神经行为功能障碍。经胡黄连苷Ⅱ干预后,缺血区神经细胞p-ERK1/2表达降低,凋亡数量减少,神经元和血脑屏障的超微结构损伤明显改善,脑梗死体积缩小,动物神经功能缺损评分减低。提示脑缺血损伤后介导神经细胞凋亡,而胡黄连苷Ⅱ可抑制神经细胞的凋亡,改善其神经组织超微结构,促进动物神经行为功能恢复。

Fig 4 Ultra-structure of blood brain barrier(EM×20 000,60 000)
A: Control Group; B: Model group; C: Treatment group; Bm: Basement membrane; Ec: Endothelial cell; Fp: Foot process; RBC: Red blood cell

Fig 5 Apoptosis in cortex of rats TUNEL×400
A: Control group; B: Model group; C: Treatment group; →indicated apoptotic cells.*P<0.05vscontrol group;#P<0.05vsmodel group
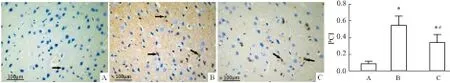
Fig 6 p-ERK1/2 expression in cortex of rats (DAB×400)
A: Control group; B: Model group; C: Treatment group. → indicated positive cells.*P<0.05vscontrol group;#P<0.05vsmodel group
Fig 7 p-ERK1/2 and ERK1/2 expressions determined by Western blot
[1] Broughton B R, Reutens D C, Sobey C G. Apoptotic mechanisms after cerebral ischemia[J].Stroke, 2009, 40(5):e331-9
[2] Kovalska M, Kovalska L, Pavlikova M, et al. Intracellular signaling MAPK pathway after cerebral ischemia reperfusion injury[J].NeurochemRes, 2012, 37(7):1568-77.
[3] Jin H, Hwang S K, Yu K, et al.A high inorganic phosphate diet perturbs brain growth, alters Akt-ERK signaling, and results in changes in cap-dependent translation[J].ToxicolSci, 2006, 90(1):221-9.
[4] Subramaniam S, Zirrqiebel U, von Bohlen Und Halbach O, et al. ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3[J].JCellBiol, 2004, 165(3):357-69.
[5] Deng X,Zhong Y, Gu L Z,et al. MiR-21 involve in ERK-mediated upregulation of MMP9 in the rat hippocampus following cerebral ischemia[J].BrainReshBull, 2013, 94(1):56-62.
[6] 李 震,徐新颖,沈 卫,等. 胡黄连苷Ⅱ对大鼠脑缺血/再灌注损伤NF-κB和I-κB的干预作用[J]. 中国药理学通报, 2010, 26(1):52-6.
[6] Li Z, Xu X Y, Shen W, et al. The interfering effects of picroside Ⅱ on the expressions of NF-κB and I-κB following cerebral ischemia reperfusion injury in rats[J].ChinPharmacolBull, 2010, 26(1): 52-6.
[7] Li X, Xu X Y, Li Z, et al. Picroside II down-regulates matrix metalloproteinase-9 expression following cerebral ischemia/reperfusion injury in rats[J].NeuralRegenRes, 2010, 5(18): 1403-7.
[8] Guo Y L, Xu X L, Li Q, et al. Anti-inflammation effects of picroside 2 in cerebral ischemic injury rats[J].BehavBrainFunct, 2010, 6(1):43-9.
[9] Li Q, Li Z, Xu X Y, et al. Neuroprotective properties of picroside II in rat model of focal cerebral ischemia[J].IntJMolSci, 2010, 11(11):4580-90.
[10]Sun L, Li X D, Wang L, et al. Anti-oxidant effect of picroside II in a rat model of cerebral ischemia/reperfusion injury[J].NeuralRegenRes, 2011, 6(15):1141-6.
[11]Naqasawa H, Kogure K. Correlation between cerebral blood flow and histologic changes in a new rat model of middle cerebral artery occlusion[J].Stroke, 1989, 20(8): 1037-43.
[12]Chen J, Sanberg P R, Li Y, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats[J].Stroke, 2001, 32(11):2682-8.
[13]赵 丽, 郭云良, 李晓丹, 等. 胡黄连苷II对脑缺血损伤后神经元特异性烯醇化酶表达的影响[J]. 中国药理学通报, 2014, 30(2): 192-9.
[13]Zhao L, Guo Y L, Li X D, et al. Effect of picroside II on neuron specific enlase after cerebral ischemic injury[J].ChinPharmacolBull, 2014, 30(2):192-9.
[14]李红云,赵 丽,宿 希,等. 胡黄连苷Ⅱ治疗脑缺血/再灌注损伤剂量和时间窗的初步探讨[J]. 中国药理学通报, 2012, 28(4): 549-53.
[14]Li H Y, Zhao L, Su X, et al. Primary study on the therapeutic dose and time window of picroside H in cerebral ischemic injury in rats[J].ChinPharmacolBull, 2012,28(4):549-53.
[15]Bhat N R, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death[J].JNeurochem, 1999, 72(1): 112-9.
[16]Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease[J].JIntMed, 2005, 258(6):479-517.
[17]Kim R, Emi M, Tanabe K, et al. Regulation and interplay of apoptotic and non-apoptotic cell death[J].JPathol, 2006, 208(3):319-26.
[18]Namura S, Iihara K, Takami S, et al. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia[J].PNAS, 2001, 98(20): 11569-74.
[19]Lobner D, Liot G. Role of MAPK/ERK in neurotrophin-4 potentiation of necrotic neuronal death[J].NeurochemRes, 2004, 29(12): 2303-9.
[20]敬乃鲁,李晓丹,赵 丽,陈红兵. 胡黄连苷Ⅱ对大鼠脑缺血损伤水通道蛋白4表达的影响[J]. 中国临床神经科学,2013,21(6):631-6.
[20]Jing N L, Li X D, Zhao L, Chen H B. The influence of picroside Ⅱ on the expression of aquaporin 4 following cerebral ischemic injury in rats[J].ChinJNeurosci, 2013, 21(6):631-6.
[21]Pei H T, Su X, Zhao L, et al. Primary study for the therapeutic dose and time window of picroside Ⅱ in treating cerebral ischemic injury in rats[J].IntJMolSci, 2012, 13(3): 2551-62.
[22]Zhao L, Guo Y L, Ji X J, et al. The neuroprotective effect of picroside II via regulating the expression of myelin basic protein after cerebral ischemia injury in rats[J].BMCNeurosci, 2014, 15(2):25-33.
Effect of picroside II on neuronal apoptosis and ultrastructure in cerebral ischemic injury in rats
WANG Ting-ting, ZHAO Li, LI Xiao-dan, ZHANG Mei-zeng, GUO Yun-liang
(InstituteofCerebrovascularDiseases,theAffiliatedHospitalofQingdaoUniversity,QingdaoShandong266003,China)
Aim To explore the effect of picroside II on neuronal apoptosis and ultrastructure after cerebral ischemic injury in rats. Methods The focal cerebral ischemic models were established by inserting a monofilament thread into middle cerebral artery occlusion (MCAO) in 60 Wistar rats and treated by injecting picroside Ⅱ (20 mg·kg-1) intraperitoneally. The neurobehavioral function was evaluated by modified neurological severity score test. The cerebral infarct volume was measured by tetrazolium chloride staining. The morphology and ultrastructure of brain tissue were observed by hematoxylin-eosin staining and transmission electron microscopy respectively. The apoptotic cells were counted by terminal deoxynucleotidyl transferase dUTP nick end labeling assay and the expression of p-ERK1/2 was determined by immunohistochemical assay and Western blot. Results The neurological behavioral malfunction and the cerebral infarct appeared in rats with MCAO. In model group, the damage of neurons and blood brain barrier (BBB) in cortex worsened, while the number of apoptotic cells and the expression of p-ERK1/2 increased more significantly than those in control group (P<0.05). In treatment group, the neurological behavioral function, morphology, ultrastructure of neuron and BBB were improved, the cerebral infarct volume, the number of apoptotic cells and the expression of p-ERK1/2 decreased more significantly than those in model group (P<0.05). Conclusion Picroside II might reduce cell apoptosis to improve the morphology and ultrastructure of cerebral ischemic tissue and recover the neurobehavioral function of rats.
picroside II; cerebral ischemia; apoptosis; ultrastructure; rats; p-ERK1/2
时间:2015-3-3 11:08 网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20150303.1108.021.html
2014-10-15,
2014-11-24
国家自然科学基金资助项目(No 81041092)
王婷婷(1988-),女,硕士生,研究方向:神经病学,E-mail: 61279652@qq.com; 郭云良(1961-),男,博士,教授,博士生导师,通讯作者,研究方向:神经病学,Tel: 0532-82991711,E-mail: guoqdsd@163.com
10.3969/j.issn.1001-1978.2015.03.021
A
1001-1978(2015)03-0400-07
R-332;R284.1;R322.81;R329.25;R743.31
