美国人道主义器械豁免政策分析
2015-06-08李少博常峰
李少博 常峰

摘要:
[目的]对比FDA的人道主义器械和传统器械审批程序,为我国完善治疗罕见病医疗器械的审批提供一定的参考。[方法]采用比较法,从申请标准、盈利限制以及上市后监察几个方面对FDA医疗器械不同的审批方式进行对比。[结果]HDE政策的实施有力推动了罕见病医疗器械的创新和发展。[结论]我国应采取激励政策,促进罕见病医疗器械的发展。
关键词:
人道主义用途器械;人道主义器械豁免;医疗器械;上市前许可
中图分类号:
D9
文献标识码:A
文章编号:16723198(2015)08018403
随着科技的不断创新,医生在工作中较之以往任何时候都更依赖于医疗器械的辅助。在过去几十年间,医疗器械产业经历了从种类、用途到复杂性的爆发式增长。目前,全球总医疗器械市场价值已超过4,118亿美元,其中近半数的产出和消耗都发生在美国。截至目前,美国共有超过6,500家医疗器械生产企业,人均医疗器械支出约为每年392美元,是全球最大的医疗器械市场。
美国食品药品监督管理局(FDA)按风险等级对医疗器械实行分类审批管理,审批方式主要有2种:上市前通告(Pre-market Notification,简称510K)和上市前审批(Pre-market Approval,简称PMA)。为了满足罕见病患者的医疗需求,美国国会于1990年颁布了《医疗器械安全法案》,提出了人道主义器械豁免(Humanitarian Device Exemption,简称HDE)的审批途径。如果某种医疗器械的目的在于治疗或诊断某种疾病或病症,且该疾病或病症每年在美国的影响人数不超过4,000名,则该器械可申请获得了人道主义用途器械(Humanitarian Use Device,简称HUD)资格,并通过HDE途径得到审评。
本文研究了FDA人道主义器械豁免途径和传统医疗器械审批途径的不同流程,着重从申请标准、盈利限制以及上市后监察等方面比较二者的差异,总结了HDE审批途径的潜在优缺点,以期为我国完善医疗器械审批方式提供一定的参考。
1FDA医疗器械的分类审批监管
1.1传统医疗器械审批流程
根据其风险等级和管理程度的不同,FDA将普通医疗器械分为I、II、III类,其中III类医疗器械的风险等级最高。对于较低风险的I类器械,FDA实施一般控制。这类器械一般无需经过上市审批,生产企业只需向FDA证明该器械符合GMP规范,并进行登记注册后即可上市销售;对于存在一定使用风险的II类器械,FDA对其实施一般&特殊管理,大多数II类器械须提交上市前通告,证明该器械与已经合法上市的器械实质性等同;对于具有较高临床风险,或是用于支持维护生命的III类器械,FDA则实行严格的上市前审批制度,器械在申请上市前须通过毒性、免疫、生物相容性试验和人体临床试验来证明其安全有效性。
对于普通医疗器械,FDA审批的具体决议流程如图1所示。
1.2人道主义用途器械审批流程
人道主义用途器械审批流程分为两个步骤,
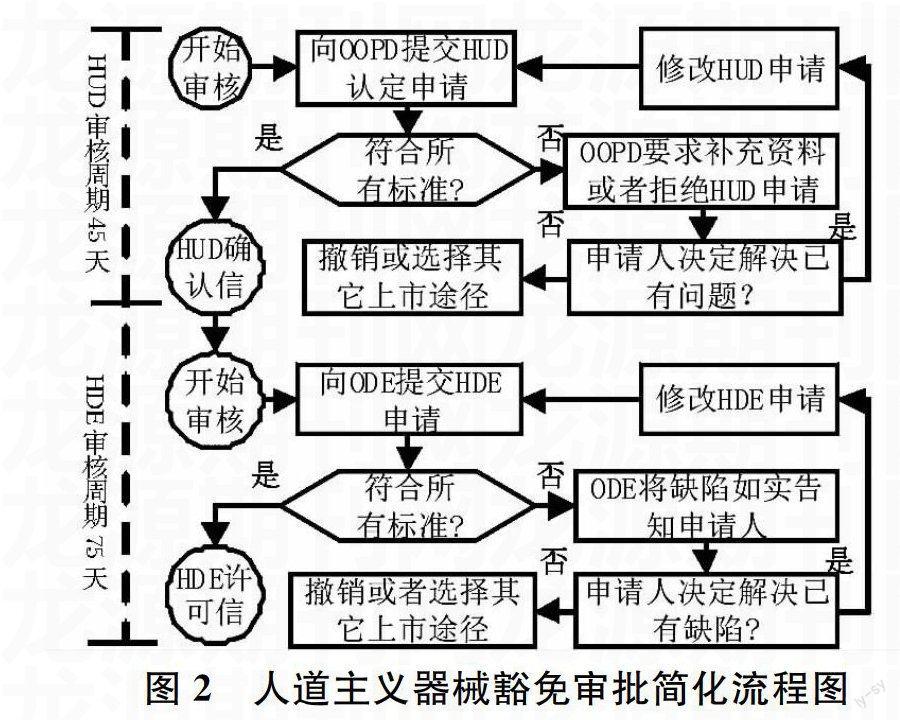
首先获得HUD确认,再获得HDE许可上市。获得HUD资格的器械须满足以下条件:(1)用于治疗或诊断患病率极低的疾病,每年在美国的影响人数少于4,000;(2)尚无其它已上市器械能够治疗该疾病;(3)使用该器械不会给患者带来严重不合理的风险;(4)可能给患者带来的益处超过疾病或损伤的风险。对于满足上述条件的器械,FDA下属的孤儿药产品开发办公室(OOPD)将在45天内发出HUD确认通知,接着FDA器械评价办公室(ODE)会审查器械的安全性、其可能带来的健康益处以及是否有可替代的器械,从而决定该器械能否获得HDE许可。该阶段的审核周期为75天。
值得注意的是,对于已获得HDE许可的器械,若后续有治疗相同适应症的医疗器械通过PMA或510K等途径上市,FDA将撤销该器械的HDE许可。
HUD的审批决议流程如图2所示。
2人道主义用途器械与传统医疗器械审批监管的对比
2.1申请标准不同
HDE审批和PMA审批的形式和内容都非常类似,二者最大的不同在于对有效性的要求。对于高风险的III类医疗器械,PMA审批往往需要通过多中心、随机临床试验,对器械预期的“安全有效性假设”加以科学验证。而HUD所针对的目标市场极为有限(每年在美国影响人数少于4,000),不可能针对这类患者做大量的随机临床试验。因此FDA规定,HUD申请中无需包含科学的临床研究证据来证明其有效性,但须证明使用该器械不会对患者构成重大伤害,同时使用它可能给患者带来的健康益处超过伤害的风险。证明可能的健康益处可参考文献资料;如果对临床数据有要求,则只需做小样本、非对照试验。
2.2上市后盈利限制规定不同
根据2002年的《医疗器械使用者付费和现代化法案》,FDA免除了HUD上市前申请的费用。但与此同时,FDA规定禁止HUD用于盈利,其售价不得高于250美元。若高于这个价格,FDA将要求企业证明其收取的费用不超过研发、制造和销售的成本。传统医疗器械则需向FDA缴纳不菲的审批费用。据FDA规定,2015财政年PMA申请的标准收费为250,895美元,510K申请的标准收费为5,018美元。对于传统医疗器械上市后的盈利,FDA并未做出任何限制。
2.3上市后使用规范不同
对于传统医疗器械,只要在医生基于合理的诊断,认为患者能从治疗中获益的情况下即可使用。而对于尚未证明有效性,仅具备较低标准安全性的HUD,明确其使用限制对于保障患者的权利和健康尤为关键。根据联邦法规,美国机构审查委员会(IRB)对HUD实施全程监管。所有HUD的使用都须经过IRB的事前审查和批准。在获得IRB批准前,HDE持有者不得向任何机构运送HUD。获得IRB许可后,HUD仅可用于治疗患有特定疾病的患者,且仅针对特定的适应症。只有在特定的紧急情形下,医生可以不经过IRB授权而先行使用,但须在5天内告知IRB。
2.4不同审批方式对比的总结
通过以上比较发现,不同审批方式存在较大差异。总结评价见表1。
3人道主义器械豁免政策的优劣势分析
3.1缩短了医疗器械的研发周期
传统的医疗器械审批方式依赖于严格的有效性研究,而临床有效性的证明需要大量的受试患者和广泛的观察终点,这就意味着研究需要耗费漫长的时间。和PMA相比,HDE申请被豁免了严格的临床研究要求,产品上市周期大为缩短,多则节省3到4年,这可能是整整一个产品生命周期的长度。例如,美敦力(Medtronic)公司治疗强迫性精神障碍的脑深部刺激系统(DBS)获得了HDE上市许可后,后续研究只需要针对较少的受试者做无对照、非随机临床试验即可。比起其他审批途径,获得HDE许可使得该产品提前了一到两年面市,在竞争极为激烈的DBS治疗精神疾病领域,美敦力无疑博得头筹。HDE因而被视为一条省时的上市捷径,也在一定程度上逆转了美国罕见病医疗器械上市的迟滞。
3.2降低了医疗器械的研发成本
传统医疗器械审批途径中,证明器械有效性所需投入的研发成本是一道无形的巨大屏障,阻碍了许多器械的上市。若企业在研发进程中获得了HDE许可,不仅可被豁免不菲的审评费用,还可通过重新设计、简化临床试验,缩减受试人数和研究年限,节省总的研发成本,从而更早的开拓产品市场,为前期的研发投入获取一定的经费补偿。比起预付大笔的研发费用的传统审批途径,这种方式对于缺乏资源的中小型企业而言更有吸引力。与此同时,企业可借助HDE途径与FDA建立良好的工作伙伴关系,提早获得医生的信任,这些因素对于产品的顺利上市都极为关键。
3.3IRB审查及盈利限制造成企业的长期负担
借助HDE审批途径,器械可能以最快的速度上市,在时间和成本方面有短期的优势,但FDA严格限定了HUD的年销售数量和售价,缺乏盈利潜力将给企业造成长期的负担。另一个负担则源于IRB繁琐的审查过程,IRB每年审查一次,企业为配合其持续审查需投入巨大精力。至少需要一名全职临床专员负责与IRB接洽,审查期间包括人事费、IRB更新费等各种支出。若企业在未获得IRB许可的前提下销售该HUD,则可能导致HDE许可被撤销的严重后果。
4讨论和建议
美国不但有着强大的医疗器械总体研发实力,在罕见病器械研发领域的成就也首屈一指。这可归功于企业追求自身利润的努力和美国政府基于人道主义援助的政策法规导向,在一定程度上也离不开监管部门的技术引导和帮助,毋庸置疑FDA的HDE政策在促进美国治疗罕见病医疗器械创新及监管方面发挥了极为重要的作用。
目前,我国罕见病领域的医疗器械研发尚属空白,临床所需的医疗器械只能依赖进口,同时许多国外已上市的HUD尚未在我国注册。我国罕见病患者的医疗需求远未得到满足,缺乏有效的治疗措施很大程度上增加了患者的发病率和死亡率。要改善目前国内研发激励严重不足的现状,加快安全有效的罕见病医疗器械的创新,不仅需要企业研发的努力,监管审评流程的相应改进也至关重要。希望我国医疗器械监管部门能给予罕见病器械相应的鼓励政策,如采取罕见病器械审批专用“绿色通道”,减免税收和基金资助等措施,在科学监管的基础上合理加快审评速度,最大限度地保证我国罕见病患者获得安全有效的医疗器械。
参考文献
[1]The Medical Device Market:USA[EB/OL].(20141212)[20150305].http://www.espicom.com/usamedicaldevicemarket.html.
[2]Maisel W H.Medical device regulation: an introduction for the practicing physician[J].Annals of Internal Medicine,2004,140(4):296302.
[3]U.S. Food and Drug Administration.Guidance for Industry and Food and Drug Administration Staff:Humanitarian Use Device(HUD) Designations[EB/OL].(20140514)[20150302].http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm366338.htm.
[4]Henderson JA,Alexander BC,Lewis AP,Smith JJ.The MDUFMA User Fee Program in 2005:A critical year[C].Center for Integration of Medicine and Innovative Technology,2005:19.
[5]Medical Device User Fees for Fiscal Year 2015[EB/OL].(2014081)[20150312].http://www.fda.gov/MedicalDevices/ResourcesforYou/Industry/ucm407660.htm.
[6]Bernad D M.Humanitarian Use Device and Humanitarian Device Exemption regulatory programs:pros and cons[J].Expert. Rev. Med. Devices,2009:137145.
