固定污染源硝酸雾中硝酸根的检测
2015-06-06王英进沈兆欣崔春梅
鲍 静,王英进,沈兆欣,崔春梅
(1. 中国运载火箭技术研究院 北京航天计量测试技术研究所,北京 100076;2. 中国运载火箭技术研究院,北京 100076)
分析与检测
固定污染源硝酸雾中硝酸根的检测
鲍 静1,王英进2,沈兆欣1,崔春梅1
(1. 中国运载火箭技术研究院 北京航天计量测试技术研究所,北京 100076;2. 中国运载火箭技术研究院,北京 100076)
建立了固定污染源硝酸雾中硝酸根的检测方法。用硅胶管采集固定污染源硝酸雾中的硝酸,用碳酸钠溶液进行洗脱,最后使用离子色谱仪测定硝酸根质量浓度。对影响检测结果的加热时间和静置时间进行了研究。实验结果表明:最佳的加热时间为10 min,无需静置;方法的相对标准偏差为0.27%~0.62%,检出限为0.002 5 μg/mL,加标回收率为99.7%~102.2%。该法线性关系良好,检出限低,精密度高,能满足固定污染源硝酸雾中硝酸根的测定要求。
硝酸雾;固定污染源;离子色谱法;分析方法
硝酸雾主要为废气中的硝酸根离子以及微粒所形成的雾。化石燃料不完全燃烧产生的氮氧化物被空气中的雾滴吸收后易氧化形成硝酸雾。根据行业内大气污染物的排放标准,许多企业需要对硝酸雾进行检测。但目前国家并未出台相关的检测标准,各检测机构未形成统一的检测方法。
废气中污染物的采样方法有直接采样法、溶液吸收法、滤膜截流法和填充柱采样法等。直接采样法主要用于高浓度污染物的测定[1]。溶液吸收法主要用于采集气态污染物[2]。滤膜截流法主要是将气体污染物拦截在滤膜上,适用于大气中气溶胶、降尘、可吸入颗粒物、烟尘等的采集,但在采集固定污染源中废气的应用领域中并不多见[3]。填充柱采样法使用不同类型的玻璃管或硅胶管采集废气,在管内填充不同的固体填充剂,用于吸附废气中的污染物。
本工作采用填充柱采样法,通过硅胶管采样,采集后的试样用碳酸钠溶液进行洗脱,使用离子色谱仪进行检测,并对分析过程中的影响因素进行了研究。
1 实验部分
1.1 试剂、材料和仪器
无水碳酸钠、硝酸钠:优级纯。
去离子水:电导率小于1 μS/cm,经0.45 μm微孔过滤膜过滤后备用。
硝酸钠贮备液:称取0.137 1 g硝酸钠,溶于去离子水中,移入1 000 mL容量瓶中定容,得到硝酸根质量浓度为1 000.0 μg/mL的硝酸钠贮备液。使用时用去离子水稀释。
碳酸钠贮备液:称取21.198 g无水碳酸钠,溶于去离子水中,移入500 mL容量瓶中定容,得到浓度为0.400 mol/L的碳酸钠溶液。使用时用去离子水稀释100倍。
硝酸雾:取自北京某显示技术有限公司工艺生产线,成分主要为硝酸。分别采集从高浓度生产线和低浓度生产线上产生的硝酸雾。
DX-120型离子色谱仪:美国戴安公司;TGB32型电光分析天平:上海天平仪器厂;GH-60E型烟气采样器:青岛金仕达仪器有限公司;22610-03型硅胶管:美国SKC公司。
1.2 试样的前处理方法
选择两种不同硝酸根质量浓度的硝酸雾进行实验。采用特定硅胶管采集固定污染源硝酸雾中的硝酸。采样前校准采样设备。采样流量为0.2~0.5 L/ min,采样时间为0.5~1 h。采样后,用5 mL浓度为0.004 mol/L的碳酸钠溶液洗脱硅胶,并在90 ℃的水浴中加热一定时间。冷却30 min后,在室温下,再加入5 mL碳酸钠溶液,混匀后静置一定时间。将试样用微孔过滤器过滤[4]。
1.3 硝酸根的测定方法
将滤液注入离子色谱仪中,测定硝酸根的质量浓度[5]。检测器为电导检测器,载气为氮气,载气流量为1 mL/min,碳酸钠溶液流量为1.5 mL/min。
2 结果与讨论
2.1 标准曲线的绘制
将一定量硝酸根质量浓度为25.0 μg/mL的硝酸钠溶液置于10 mL比色管中,加入浓度为0.004 mol/ L的碳酸钠溶液定容,得到一系列不同硝酸根质量浓度的溶液。以硝酸根质量浓度为横坐标,峰高为纵坐标绘制标准曲线,标准曲线见图1。标准曲线方程为y=27.168x-1.788 5,相关系数为0.999 8。

图1 标准曲线
2.2 加热时间的确定
试样的解吸程度与加热时间有关。在静置时间为0的条件下,加热时间对硝酸根质量浓度的影响见表1。由表1可见,随加热时间的延长,硝酸根质量浓度呈现出一定的上升趋势,但变化趋势很小。综合考虑采集试样的充分解吸及实验时间的节省,选择加热时间为10 min较适宜。

表1 加热时间对硝酸根质量浓度的影响
2.3 静置时间的确定
在加热时间为10 min的条件下,静置时间对硝酸根质量浓度的影响见表2。由表2可见,延长静置时间,并没有更多的硝酸根溶解到碳酸钠溶液中。由此可见,试样在碳酸钠溶液中的解吸过程进行很快。因此,从减少时间成本考虑,选择无需静置直接进行后续实验。

表2 静置时间对硝酸根质量浓度的影响
2.4 精密度
对两种不同硝酸根质量浓度的硝酸雾试样分别进行6次平行测定,精密度计算结果见表3。由表3可见,该方法的相对标准偏差为0.27%~0.62%,精密度高于余波等的硝酸雾检测方法(相对标准偏差为2.4%)[2]。

表3 精密度计算结果
2.5 检出限
按照HJ 168—2010《环境监测 分析方法标准制修订技术导则》[6]中的公式来计算方法检出限,见式(1)。

式中:MDL为方法检出限, μg/mL;n为试样的平行测定次数;t(n-1,0.99)为自由度为n-1、置信度为99%时的t分布(单侧);S为n次平行测定的标准偏差, μg/mL。
重复7次空白实验,计算7次平行测定的标准偏差为0.000 8 μg/mL。查t分布表可知t(6,0.99)= 3.143。计算得到该方法的检出限为0.002 5 μg/mL,远低于HJ/T 84—2001《水质 无机阴离子的测定 离子色谱法》[7]中给出的水中硝酸根检测方法的检出限(0.03 μg/mL)。
2.6 加标回收率
向空白硅胶管中分别加入硝酸根质量浓度为2.000 μg/mL和10.000 μg/mL的硝酸钠溶液进行加标回收实验,加标回收率计算结果见表4。由表4可见,该方法的加标回收率为99.7%~102.2%。
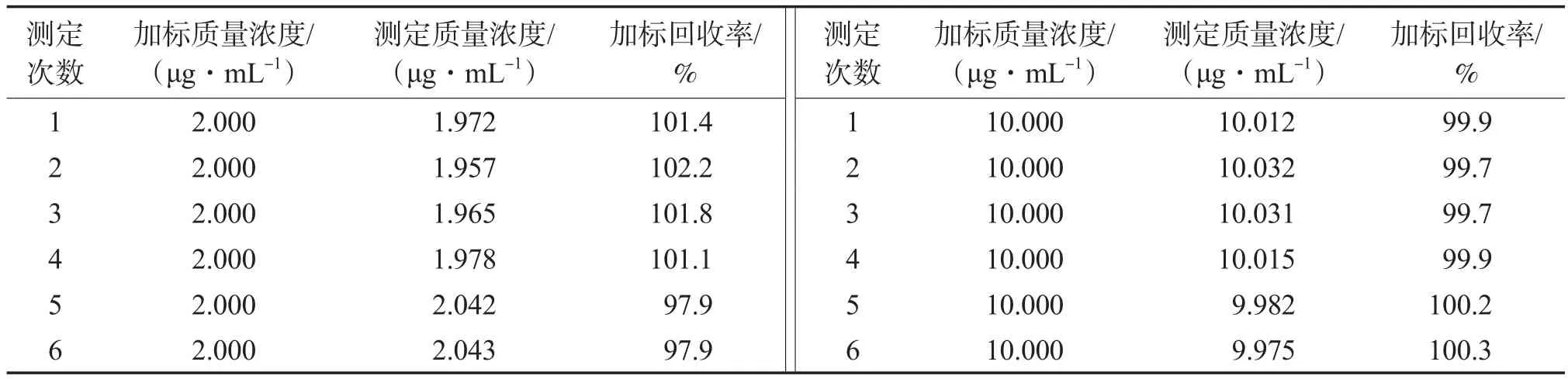
表4 加标回收率计算结果
3 结论
a) 建立了固定污染源中硝酸雾的检测方法。用硅胶管采集固定污染源硝酸雾中的硝酸,用碳酸钠溶液对采集后的试样进行洗脱,最后使用离子色谱仪进行检测。
b) 对影响检测结果的加热时间和静置时间进行了研究。实验结果表明, 最佳的加热时间为10 min,无需静置。
c) 该方法的相对标准偏差为0.27%~0.62%,检出限为0.002 5 μg/mL,加标回收率为99.7%~102.2%。采用该方法检测固定污染源硝酸雾中的硝酸根,效果好,线性关系良好,检出限低,精密度高。
[1] 王黎伟,蒲凤莲. 污染源废气监测过程采样解析[J].能源环境保护,2007,21(2):54 - 56.
[2] 余波,沈锴. 离子色谱法测定固定污染源排气中的氯化物和硝酸盐[J]. 浙江科技学院学报,2003,15(增刊):113 - 114.
[3] 茅海琼. 超声波萃取离子色谱法测定固定污染源有组织废气中的硫酸雾[J]. 分析仪器,2011(2):30 -31.
[4] 赵淑岚,张健,李梅莉,等. 工作场所空气中四种阴离子的离子色谱测定法[J]. 中华劳动卫生职业病杂志,2006,24(10):617 - 620.
[5] 《空气和废气监测分析方法》编委会. 空气和废气监测分析方法[M]. 4版. 北京:中国环境科学出版社:450 - 452.
[6] 环境保护部科技标准司. HJ 168—2010 环境监测 分析方法标准制修订技术导则[S]. 北京:中国环境科学出版社,2010.
[7] 国家环境保护总局科技标准司. HJ/T 84—2001 水质无机阴离子的测定 离子色谱法[S]. 北京:中国环境科学出版社,2001.
(编辑 王 馨)
Determination of Nitrite in Nitric Acid Fog from Stationary Source Emission
Bao Jing1,Wang Yingjin2,Shen Zhaoxin1,Cui Chunmei1
(1. Beijing Aerospace Institute for Metrology and Measurement Technology,China Academy of Launch Vehicle Technology,Beijing 100076,China;2. China Academy of Launch Vehicle Technology,Beijing 100076,China)
The method for determination of nitrite in nitric acid fog from stationary source emission was established. Nitric acid was collected from the nitric acid fog using a silicon tube,and then was eluted with sodium carbonate solution,in the end was detected by ion chromatography. The effects of heating time and standing time on determination were studied. The experimental results show that:The best heating time is 10 min and the standing time is 0;The relative standard deviation of the method is 0.27%-0.62%,the detection limit is 0.002 5 μg/mL,the recovery of standard addition is 99.7%-102.2%. This method can meet the requirement for determination of nitrite in nitric acid fog from stationary source emission with good linearity,low detection limit and high precision.
nitric acid fog;stationary pollution source;ion chromatography;analysis method
X831
A
1006 - 1878(2015)01 - 0103 - 04
2014 - 08 - 10;
2014 - 11 - 17。
鲍静(1985—),女,安徽省安庆市人,硕士,工程师,电话 13501134967,电邮 baojing850324140@163.com。
