嵌段共聚物直接热解法制备介孔炭材料及其在超级电容器中的应用
2015-06-05孔令斌李晓明罗永春
汪 勇, 孔令斌,, 李晓明, 冉 奋, 罗永春, 康 龙
嵌段共聚物直接热解法制备介孔炭材料及其在超级电容器中的应用
汪 勇1, 孔令斌1,2, 李晓明1, 冉 奋1, 罗永春2, 康 龙2
(1.兰州理工大学省部共建有色金属先进加工与再利用国家重点实验室,甘肃兰州730050; 2.兰州理工大学材料科学与工程学院,甘肃兰州730050)
以嵌段共聚物为前驱体,通过直接热解聚丙烯腈嵌段苯乙烯(PAN-b-PS-b-PAN)制备新型纳米多孔炭材料。炭材料制备依赖于嵌段共聚物分子的设计,而分子量可控、分布范围较窄的嵌段共聚物则通过可逆加成链转移(RAFT)聚合方法合成。所制炭材料不仅具有较高的比表面积(950 m2·g-1),且在2~4 nm的介孔范围内孔径得到良好的控制。此外,作为电极材料在2 mol/L KOH电解液中表现出高的比容量(185 F·g-1,电流密度为0.625 A·g-1),且显示较好的循环寿命,经10 000次循环后,能够保持初始比容量的97.5%。通过不同分子量聚合物的设计,制备结构新颖的多孔炭材料,可应用于高性能超级电容器。
嵌段共聚物;介孔炭;聚合物炭化;能量存储;超级电容器
1 Introduction
Supercapacitors play a key role in supporting the voltage of a system during increased loads from portable equipments to electric vehicles[1].Supercapacitors has been expected as a secondary electric power supplier for the automobiles with hybrid engine or fuel cell motor because of its rapid charge and discharge[2].Furthermore supercapacitor offer extremely a long service life over 106cycles,a high specific power of more than 10 Kw·h·Kg-1and can now be designed on the basis of environmentally friendlycomponents[3].Currently,Supercapacitors are widely used in consumer electronics[4],memory back-up systems,industrialpower and energy management[5], which can alleviate shortcomings of batteries,such as slow energy discharge.However,the energy density of supercapacitors is smaller than that of secondary batteries,which is a drawback for practical devices. In order to make the supercapacitors widely used in mang fields,the development of supercapacitors with a high energy density is highly needed.
In general,carbons are good electrode materials of supercapacitors[6],which have been widely used and extensively studied,owing to their high conductivity,high surface area(up to 2 000 m2·g-1)[7], excellent anti-corrosion property,controllable porous structure and relatively low cost[8].Up to now,the preparation methods of carbon material for supercapacitors are reported extensively.For example,CMK-3 derived from the hard template SBA-15[9]shows a fiberlike close stacking structure with a pore diameter of 3.9 nm and a surface area of 900 m2·g-1,and a specific capacitance of 10 mF·cm-2or 90 F·g-1in nonaqueous electrolyte[10].Recently,a direct synthesis of highly ordered mesoporous carbons has been achieved by utilization of the organic-organic co-assembly between the commercial amphiphilic Pluronic block copolymers and phenolic oligomers through an evaporation induced self-assembly or solution synthesis route[11].This soft-templating synthesis procedure has great advantages over the complicated hard-templating procedures,but there are few choices for soft template.However,both the soft and hard template methods are difficult to prepare large-pore ordered mesoporous carbons due to the limitation of molecular weight of the templates’[11,12].The unique self-assembling properties of block copolymers make it possible to prepare carbon materials with tunable sizes on the nanometer scale.
Herein we demonstrate a synthesis routine of polyacrylonitrile-b-polystyrene-b-pdyacrylonitrile (PAN-b-PS-b-PAN)block-copolymer as a carbon precursor of mesoporous carbon for supercapacitors with a reversible addition-fragmentation chain transfer (RAFT)mechanism.During high temperature carbonization,styrene is released to generate mesopores, which is named as“sacrificial”segment,while polyacrylonitrile is converted partly into carbon framework (“reserved”segment).In the bulk,block copolymers undergo micro-phase separation on the molecular scale(20-200 nm)to produce complex nanostructures with various morphologies,depending on the relative volume fraction of PAN to PS.In this work,a wellcontrolled the pore structure is realized by adjusting the molecular weight of the“sacrificial”segment. Meanwhile,mesoporous carbons have been synthesized with pore sizes of 2-4 nm.The studies shows that as-prepared mesoporous carbon has a considerably high specific capacitance of 185 F·g-1at a current density of 0.625 A·g1and excellent cycle stability of~97.5%of initial capacitance after 10 000 cycles in 2 mol/L KOH aqueous solution.All the electrochemical tests demonstrate that as-prepared porous carbon possesses excellentelectrochemical performance.
2 Experimental
2.1Materials
All reactions were carried out in nitrogen atmosphere.Petroleum ether(30-60℃),chloroform (CHCl3),acetone,carbon disulfide(CS2),tetrabutyl ammonium hydrogen sulfate(TBAHS, C16H37NO4S,99%,Fluka),methanol(CH2OH), acrylonitrile(AN),styrene(St),N,N-dimethylformamide(DMF)were supplied by Sinopharm Chemical Reagent Co.(China)and other chemicals were of analytical grade.All chemicals were puriɦed by distillation.Azodiisobutyronitrile(AIBN)was purified by recrystallization in ethanol at 40℃.
2.2Preparation of chain transfer agent
Synthesis of S,S'-Bis(α,α'-dimethyl-α″-acetic acid)-trithiocarbonate(BDMAT)was performed according to document[13].
2.3Preparation of PAN macroinitiator
In a typical synthesis,for PAN synthesis,1× 10-1g(7.09×10-3mol)of BDMAT,3.2×10-2g (3.9×10-3mol)of AIBN,30 mL of DMF,and 10 g of acrylonitrile were added into an eggplant-bottom flask with a magnetic stirring bar,which was closed with stopcocks,subjected to three freeze-pump-thaw cycles,and immersed into a thermostated water bath at80℃for12 h under stirring.The reaction was terminated by the addition of aerated DMF,then the product was precipitated by adding methanol.The solid products was dried in a vacuum oven at room temperature.
2.4Preparation of block copolymer PAN-b-PS-b-PAN
For the synthesis of PAN-b-PS-b-PAN block copolymer,the above synthesized PAN(Mn= 12 203 g·mol-1and MW/Mn=1.09)was used as a macroinitiator.Feed ratios were calculated according to the proportion of PS:PAN-BDMAT:AIBN= 1 000∶3.7∶1 for the copdymer 1 and PS∶PAN-BDMAT∶AIBN=1 000∶1∶1 for the copolymer 2,and 30 mL DMF were added into an eggplant-bottom flask with a magnetic stirring bar,and the flask was closedwith stopcocks and subjected to three freeze-pumpthaw cycles,immersed into a thermostated water bath at 80℃for 12 h with stirring.The reaction was terminated by the addition of aerated DMF,the products were precipitated by adding methanol.The solid products were dried in a vacuum oven at room temperature to obtain block copolymer 1and 2.
2.5Preparation of mesoporous carbons
The polymers,PAN or the block copolymers were heated in air at 20℃·min-1to 240℃for 6 h. After pre-oxidation and cross-linking of polymers,the samples were pyrolyzed at the 800℃at a heating rate of 1℃·min-1in a nitrogen atmosphere to allow the conversion of the polymer to carbon materials.The pyrolyzed product from PAN was labeled as PAC, and those from PAN-b-PS-b-PAN copolymer 1and 2 were labeled as PASC-1 and PASC-2.
2.6Electrode preparation and electrochemical measurements
The working electrodes were prepared as follows:80%of electroactive material powder was mixed with 7.5%of acetylene black and 7.5%of conducting graphite in an agate mortar until a homogeneous black powder was obtained.To this mixture, 5%of tetrafluoroethylene was added together with a few drops of ethanol.After the solvent is briefly evaporated,the resulting paste was pressed at 10 MPa to nickel foam current collector.The electrode was dried at60℃for 12 h.
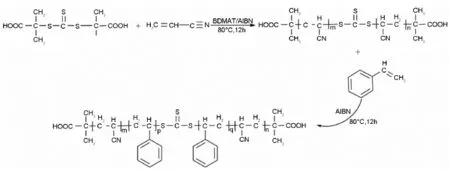
Scheme 1 Synthetic route of polyacrylonitrile-b-polystyrene-b-polyacrylonitrile block copolymers.
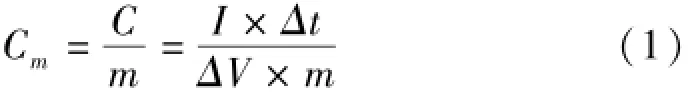
Where Cm(F·g-1)is the specific capacitance,I (A)is discharge current,Δt(s)is the discharge time,ΔV(V)represents the potential drop during discharge process,and m(g)is the mass of the active material.
2.7Structure characterization
The1H-nuclear magnetic resonance(NMR) spectra were performed in DMSO-d6with a Bruker Varian(FT-80A)NMR instrument.The molecular
All electrochemical measurements of as-prepared electrode were carried out using an electrochemical working station(CHI660C,Shanghai,China)in a half-cell setup configuration at room temperature and a 2 mol/L KOH solution as the electrolyte.A typical three-electrode glass cell equipped with a working electrode,a platinum foil counter electrode,and a saturated calomelreference electrode(SCE)was used for electrochemical measurements of as-prepared working electrodes.The corresponding specific capacitance was calculated from the equation(1): weight of the polymers was investigated using Gel Permeation Chromatography(GPC)using a Waters 1515 HPLC equipped with a Waters 2414 Refractive Index detector and waters styragel columns(500, 103,and 105).DMF(flow rate of 1.0 mL·min-1) was used as the solvent and polystyrene(Shodex standard)as the standard for a universal calibration. Hermo gravimetric analysis(TGA)and differential scanning calorimetry(DSC)were carried outin air at a heating rate of 10℃·min-1on a NETZSCH STA 449F3.The microstructures of these carbons were characterized by transmission electron microscope (TEM,JEOL,JEM-2010,Japan)and and field emission scanning electron microscope(SEM,JEOL, JSM-6701F,Japan).The characteristic functional groups of the polymers were also analyzed with a Fourier transform infrared(FT-IR)spectrometer (NEXUS 670 FT-IR)in the absorbance mode (4 500-500 cm-1;number of scans:64;resolution: 2 cm-1).The surface area and N2adsorption-desorption isotherms were measured at77 K on a Micromeritics(ASAP,2020)according to the Brunaurer-Em-mett-Teller(BET)method,the pore size distributions were derived from the desorption branches of the isotherms.
3 Results and discussion
3.1Synthesis of PAN-b-PS-b-PAN block copolymer
The1H NMR spectrum in Fig.1 shows that the signals at 1.39 ppm is assigned to-CH3of the BDMAT.The signals of the PAN block corresponding to -CH2and-CH protons are observed at 2.0 and 3.3 ppm,respectively.The two signals at 6.2-7.5 ppm are attributed to the meta-and para-protons whereas 8.0 ppm is contributed by ortho protons of benzene ring.To further elucidate PAN-b-PS-b-PAN,FTIR was employed to measure the functional groups of the copolymer materials.

Fig.11H NMR spectrum of PAN-b-PS-b-PAN block copolymer in DMF solvent.
The IR spectra of PAN-BDMAT and block copolymer PAN-b-PS-b-PAN exhibit well-defined peaks that are characteristic of the two polymers (Fig.2).The characteristic adsorption peak of the copolymer PAN is located at 2 240 cm-1,which is due to the bond C≡ N.Strong peaks near 2 929 and 1 492 cm-1indicate the stretching and bending vibrations of C—H,respectively[14,15].The BDMAT shows its characteristic absorption at 1 675 and 1 093 cm-1,which corresponds to the bonds C= O and C= S,respectively.Furthermore,the bands at 1 249 and 1 384 cm-1correspond to C—O stretching and the-C(CH3)2groups[16].It is clearly observed by a comparison of PAN with the block copolymer that there are some new absorption peaks.The additional band obtained at 3 027 cm-1is mainly caused by the C—H stretching vibration of aromatic functional group,which also has a weak bending vibration mode corresponding to the band recorded at 1 452 cm-1[17].Meanwhile,the vibrations of benzene ring skeleton C-C is located at around 1 600 cm-1[18].In the block copolymer,the characteristic vibration bands of polystyrene appear,which indicates that the block copolymers are synthesized.

Fig.2 FT-IR spectra of(a)PAN-BDMAT and(b)block copolymer PAN-b-PS-b-PAN.
Two differentblock copolymers with a controlled molecular weight and narrow polydispersity(<1.3) are listed in Table 1.The block copolymer sharply shifted to a high molecular weight region with a narrow molecular weight distribution.The molecular weight of the PS block is controlled from 12 203 to 20 086 g·mol-1with a narrow polydispersity(PDI<1.2)while the molecular weight of PS block is controlled from 12 200 up to 32 300 g·mol-1,which is expected to form large pore.

Table 1 Molecular weight of PAN-b-PS-b-PAN block copolymers by a simple polymerization process in the presence of the RAFT agent.
3.2Mesoporous carbon from block copolymer and electrochemical performence
The weightloss associated with pyrolysis is studied by TGA.Fig.3 shows weight changes during oxidative stabilization and pyrolysis of copolymers.The PAN-BDMAT shows a 5.2%of the weight loss at 270℃in a nitrogen atmosphere,and a total weight loss of 52.5%upon pyrolysis heating to 1 000℃, while the pristine and purified block copolymer exhibit a total 77%weight loss after pyrolysis 1 000℃. The weight loss of the block copolymer is clearly observed during heating process,which takes place roughly in three stages noted by stage I,II,and III. The first stage corresponds to due to partial dehydrogenation and cross-linking in the range of 20-270℃, which is the stabilization step involving the intra and intermolecular reactions that are needed to stabilizethe microphase separation structure prior to pyrolysis[19].The second stage of weight loss 59%can be clearly observed by TGA in the temperature range from 270 to 480℃.The major weightloss at450℃can be assigned to the thermal decomposition of sacrificial PS block.The third mass loss of 6%is attributed to further dehydrogenation and partialdenitrogenation of PAN.
Numerous macropores and mesopores are evidentin Fig.4b,which are in favor of the improvement in the power performance capability of resulting supercapacitor.It is well documented that macropores serve as ion buffering reservoirs and mesopores are capable of overcoming the primary kinetic limits of electrochemical process.Fig.4 c exhibits numerous mesopores with uniform pore size distribution of 2-4 nm.Thus,the prepared sample is considered to be a interconnected porous carbon.

Fig.3 TGA curves of(a)PAN-BDMAT and(b)PAN-b-PS-b-PAN.

Fig.4 (a)SEM images of PASC-2.(b)TEM of PASC-2.(c)HRTEM of PASC-2.
Nitrogen sorption experiments are performed to evaluate the overall porosity of the carbon samples (Fig.5a).Both of the PASC-1 and PASC-2 samples show type-IV isotherms with narrow hysteresis loops according to the IUPAC classification[20].The N2uptake of the PAC atlow relative pressures(p/p0<0.1) indicates the presence of the micropores,which is formed by pyrolysis of PAN.The PAC from PAN has a pore volume of0.21 cm3·g-1.The pore volume of the mesoporous carbons from the block polymers are larger(0.53,0.78 cm3·g-1)than that of porous carbon from PAN.The PAC shows a BET surface area of 467.8 m2·g-1while those of PASC-1 and-2 are 896.12 and 953.53 m2·g-1.Moreover,the pore size distribution curves of PAC(Fig.5 b)suggested that the size ofthe mesopores is about2.0 nm and virtually no other pores.PASC-1 and-2 show similar pore size distributions at2 nm[21],and the dominant pores are mesopores(>2 nm).The large mesoporous structure(6-200 nm)(insetin Fig.5b)is formed by interparticle voides from micro phase separation of the block coploy mers.In our case,the t-plot analysis shows thatthe ratio of pores(micropores and mesopores)is determined by ratio of retention block to sacrificial block of polymeric precursor.The one relatively with larger molecular weight and low polydispersity of sacrificial block,not only generated the micropores and mesopores,but also the larger distribution range(6-200 nm).These results indicate an excellent porosity of the mesoporous carbon by pyrolysis of polymers,including high surface area,large pore volume,and uniform mesopores,which are important for wide applications.
Fig.6a shows the CV curves of PAC,PASC-1 and PASC-2 samples at a scan rate of 5 mV·S-1between-1.0 V and 0 V in 2 mol/L KOH solution. The shape of the CV reveals thatthe capacitance characteristic is very similar to thatof electric double-layer capacitance in which the shape is normally close to an ideal rectangular shape,and no obvious faradaic current is observed in the voltammogram[22].The PAC exhibits a small rectangular curve,corresponding to a low capacitance.The high currentresponse in the CV curve of PASC-1 and PASC-2 indicates thatthe effective specific surface area contributing to the capacitance is remarkably improved in the carbon materials.
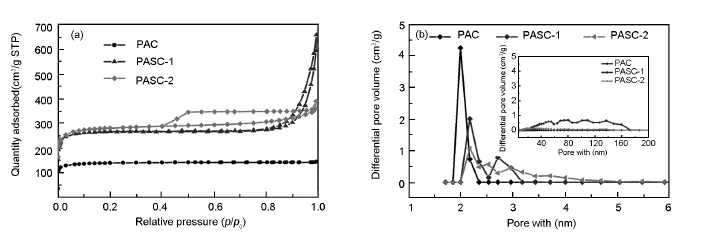
Fig.5 (a)Nitrogen sorption isotherms of PAC,PASC-1 and PASC-2 porous carbons synthesized via PAN-b-PS-b-PS block polymer pyrolysis. (b)Pore size distributions are calculated by BJH method from the desorption branches. The inset shows their corresponding pore size distribution from 20 to 200 nm.

Fig.6 Comparison of(a)cyclic voltammograms of PAC,PASC-1 and PASC-2 at a scan rate of 5 mV·s-1in 2 mol/L KOH aqueous solution; (b)Charge-discharge curves of PAC,PASC-1 and PASC-2 at a scan rate of 5 mV·s-1; (c)Complex-plane impedance plots of PAC,PASC-1 and PASC-2; (d)Specific capacitance of as-prepared samples at a controlled current densities.The mass of active material is 8 mg.
Fig.6b further displays the comparison of galvanostatic charge-discharge curves of PAC,PASC-1 and PASC-2,which have nearly triangular shapes at the same current density of 0.625 A·g-1.The mass specific capacitance(F·g-1)at the current density of 0.625 A·g-1tends to increase with the increase of specific surface area,which may be attributed to the relative narrow distribution of pore size and the increased the effective surface area.Fig.6c shows the impedance plot of PAC,PASC-1 and PASC-2.The impedance plots for each sample can be divided into a high-frequency component and a low-frequency component.The absence of this loop indicates that the materials have a really low intrinsic resistance which includes the total resistances of the ionic resistance in the electrolyte,the intrinsic resistance of active materials,and the contact resistance at the active material/ current collector interface.The semicircle in the high-frequency range associates with the surface properties of the porous electrode,which corresponds to the charge transfer resistance(Rct).It can be seen that the sample of PASC-2 have a small Rctwhich is ascribed to the nanoporous structure feature that facilitate the transport of electrolyte ions.In Fig.6d,the variation in the specific capacitance of the electrodes as a function of the scan rates is plotted.The PASC-2 exhibits the highest specific capacitance of 185 F·g-1at a current density of 0.625 A·g-1.When the scan rate is increased by ten times,the mass specific capacitance was 125 F·g-1,which is 67.5%of the initial capacity(at a current density of 0.625 A·g-1).
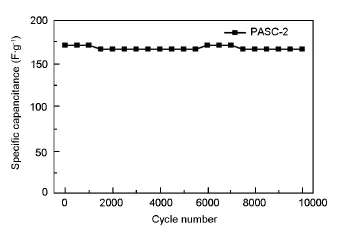
Fig.7 Cycle life at a current density of 2 A·g-1.
For a further understanding of the electrochemical performance,the long-term cycle stability of the electrode was evaluated at a current density of 2 A·g-1for 10 000 cycles.Importantly,studied devices exhibites good cycle stability,which is especially desirable in supercapacitors,by maintaining 97.5%of capacitance after 10 000 charge/discharge cycles.The long-term stability implies an excellent electrode materialfor supercapacitor.
3 Conclusions
We successfully synthesized PAN-b-PS-b-PAN block copolymers as a new kind of precursors for the formation of mesoporous carbon materials via a reversible addition-fragmentation chain transfer mechanism,in which PAN is the carbon source that foams carbon network and PS is the sacrificial segment that is released during pyrolysis at 800℃.The mesoporous carbons exhibited high BET surface areas of 896.12 m2·g-1(PASC-2)and 953.53 m2·g-1(PASC-1)with mesopore size of 2-4 nm and small amount of micropores.Moreover,the specific capacitances of PASC-2 and PASC-1 are 185 and 150 F·g-1at a current density of 0.625 A·g-1respectively and long cycle life of 97.5%capacity retention after 10 000 charge/discharge cycles in 2 mol/L KOH aqueous solution.
[1] Xiao H,Wen C,Ni C,et al.Effectof porosity on the electrical resistivity of carbon materials[J].New Carbon Materials,2013, 28(5):349-354.
[2] Mitani S,Lee S I,Yoon S H,et al.Activation of raw pitch coke with alkali hydroxide to prepare high performance carbon for electric double layer capacitor[J].Journalof Power Sources, 2004,133(2):298-301.
[3] Broussea T,Toupina M and Bélangera D.A hybrid activated carbon-manganese dioxide capacitor using a mild aqueous electrolyte[J].Journal of The Electrochemical Society,2004,151 (4),A614-A622.
[4] Zheng J P.Theoretical energy density for electrochemical capacitors with intercalation electrodes[J].Journal of The Electrochemical Society,2005,152(9):A1864-A1869.
[5] Li WR,Chen D H,Li Z,etal.Nitrogen-containing carbon spheres with very large uniform mesopores:The superior electrode materials for EDLC in organic electrolyte[J].Carbon,2007,45(9):1757-1763.
[6] Chen C M,Zhang Q,Huang C H,et al.Macroporous‘bubble’graphene film via template-directed ordered-assembly for high rate supercapacitors[J].Chemical Communication,2012,48,7149-7151.
[7] Zheng J P and Jow T R.A new charge storage mechanism for electrochemical capacitors[J].Journal of The Electrochemical Society,1995,142(1):L6-L8.
[8] Pandolfo A G and Hollenkamp A F.Carbon properties and their role in supercapacitors[J].Journalof Power Sources,2006,157 (1):11-27.
[9] Liu H J,Cui W J,Jin L H,et al.Preparation of three-dimensional ordered mesoporous carbon sphere arrays by a two-step templating route and their application for supercapacitors[J]. Journal of Materials Chemistry,2009,19:3661-3667.
[10] Li H Q,Luo J Y,Zhou X F,et al.An ordered mesoporous carbon with short pore length and its electrochemical performances in supercapacitor applications[J].Journal of The Electrochemical Society,2007,154(8):A731-A736.
[11] Deng Y H,Cai Y,Sun Z K,et al.Controlled synthesis and functionalization of ordered large-pore mesoporous carbons[J]. Advanced Functional Materials,2010,20(21):3658-3665.
[12] Liu C,Li F,Ma L P,etal.Advanced materials for energy storage[J].Advanced Functional Materials,2010,22(8):E28-E62.
[13] Lai J T,Filla D and Shea R.Functional polymers from novel carboxyl-terminated trithiocarbonates as highly efficient RAFT agents[J].Macromolecules,2002,35(18):6754-6756.
[14] Chen J Z,Chen Z H,Wang C H,et al.Calcium-assisted hydrothermal carbonization of an alginate for the production of carbon microspheres with unique surface nanopores[J].Materials Letters,2012,67(1):365-368.
[15] Liao Y H,Zhou D Y,Rao M M,et al.Self-supported poly (methyl methacrylate-acrylonitrile-vinyl acetate)-based gel electrolyte for lithium ion battery[J].Journal of Power Sources, 2009,189(1):139-144.
[16] Wang X X,Li T H,Ji Y B,etal.Synthesis and characteristicsof continuous mesoporous carbon films by a rapid solvent evaporation method[J].Applied Surface Science,2008,255(5): 1719-1725.
[17] Ho H T,Martin E L,Pascual S,etal.Thermoresponsive block copolymers containing reactive azlactone groups and their bioconjugation with lysozyme[J].Polymer Chemistry,2013,4 (3):675-685.
[18] Tan Y T,Ran F,Wang L R,et al.Synthesis and electrochemical properties of hollow polyaniline microspheres by a sulfonated polystyrene template[J].Applied Polymer,2013,127(3): 1544-1549.
[19] Zhong MJ,Kim E K,McGann J P,et al.Electrochemically active nitrogen-enriched nanocarbons with well-defined morphology synthesized by pyrolysis of self-assembled block copolymer[J]. Journal of American Chemical Society,2012,134(36):14846-14857.
[20] Zhang J,Kong L B,Cai J J,etal.Hierarchically porous nickel hydroxide/mesoporous carbon composite materials for electrochemical capacitors[J].Microporous and Mesoporous Materials,2010,132(1-2):154-162.
[21] Nguyena Chi-thanh,Kim Dong-pyo.Direct preparation of mesoporous carbon by pyrolysis of poly(acrylonitrile-b-methylmethacrylate)diblock copolymer[J].Journal of Materials Chemistry, 2011,21,14226-14230.
[22] Lang J W,Yan X B,Yuan X Y,et al.Study on the electrochemical properties of cubic ordered mesoporous carbon for supercapacitors[J].Journal of Power Sources,2011,196(23): 10472-10478 .
Mesoporous carbons for supercapacitors obtained by the pyrolysis of block copolymers
WANG Yong1, KONG Ling-bin1,2, LI Xiao-ming1, RAN Fen1, LUO Yong-chun2, KANG Long2
(1.StateKeyLaboratoryofAdvancedProcessingandRecyclingofNonferrousMetals,SchoolofMaterialsScienceandEngineering,LanzhouUniversityofTechnology,Lanzhou730050,China;
2.SchoolofMaterialsScienceandEngineering,LanzhouUniversityofTechnology,Lanzhou730050,China)
Novel mesoporous carbons were prepared by the simple pyrolysis of block-copolymers,polyacrylonitrile-b-polystyreneb-polyacrylonitrile(PAN-b-PS-b-PAN),in which PAN generates a carbon network and PS is released to form mesopores after pyrolysis.The block-copolymers were synthesized by reversible addition-fragmentation chain transfer(RAFT)polymerization and the molecular weight of each polymer block can be designed to tailor the porous structure of the mesoporous carbons.The carbons have high specific surface areas and a well-controlled mesopore size.The best mesoporous carbon has a high specific capacitance of 185 F ·g-1at0.625 A·g-1with a high power capability and a remarkable cycle stability in 2 mol/L KOH aqueous electrolyte.
Block copolymer;Mesoporous carbon;Polymer carbonization;Energy storage;Supercapacitor
KONG Ling-bin,Professsor.E-mail:konglb@lut.cn
TQ127.1+1
A
国家自然科学基金(51362018,21163010);教育部关键项目(212183).
孔令斌,教授.E-mail:konglb@lut.cn
汪 勇,硕士研究生.E-mail:gaoxing9999@126.com
1007-8827(2015)04-0302-08
Received date:2015-01-05;Revised date:2015-07-25
Foundation item:National Natural Science Foundation of China(51362018,21163010);Key Project of Chinese Ministry of
Education(212183).
Author introduction:WANG Yong,Master Student.E-mail:gaoxing9999@126.com
English edition available online ScienceDirect(http://www.sciencedirect.com/science/journal/18725805).
10.1016/S1872-5805(15)60191-3
