TGFB1基因突变导致罕见进行性骨干发育不良
2015-06-01徐晓杰马豆豆王建一夏维波邢小平
徐晓杰,马豆豆,吕 芳,刘 怡,王建一,姜 艳,王 鸥,夏维波,邢小平,李 梅
中国医学科学院 北京协和医学院 北京协和医院内分泌科卫生部内分泌重点实验室,北京100730
TGFB1基因突变导致罕见进行性骨干发育不良
徐晓杰,马豆豆,吕 芳,刘 怡,王建一,姜 艳,王 鸥,夏维波,邢小平,李 梅
中国医学科学院 北京协和医学院 北京协和医院内分泌科卫生部内分泌重点实验室,北京100730
目的 分析1个进行性骨干发育不良 (progressive diaphyseal dysplasia,PDD)家系患者的临床表型,并检测转化生长因子β1编码基因TGFB1的突变类型。方法 1例幼年起病,表现为下肢骨痛、无力和肌肉减少的PDD患者,来自非近亲婚配家庭,评估其临床表现、骨骼X线特点、骨转换生化指标水平;采用聚合酶链式反应及其产物直接Sanger测序法检测TGFB1突变。结果 患者骨转换水平增高,影像学提示患者四肢骨皮质不均匀性增厚、硬化。基因检测提示患者TGFB1基因第4外显子存在c.652C>T杂合性错义突变 (p.Arg218Cys),患儿父母均未发现该突变。予患者糖皮质激素治疗,治疗4个月后患者骨痛缓解、活动能力明显改善。结论 四肢骨痛和骨干皮质增厚是PDD的典型临床表现,TGFB1第218位点错义突变为PDD热点致病突变类型,糖皮质激素治疗能够缓解PDD病情。
进行性骨干发育不良;表现型;TGFB1基因;突变
Med J PUMCH,2015,6(5):327-332
进行性骨干发育不良 (progressive diaphyseal dysplasia,PDD,OMIM 131300),又称 Camurati-Engelmann病 (Camurati-Engelmann disease,CED),是一种以长骨骨干进行性、对称性皮质增厚为特点的罕见遗传性骨骼疾病,患病率为百万分之一[1]。该病呈常染色体显性遗传,其致病基因TGFB1位于19q13,该基因激活性突变导致其编码蛋白转化生长因子 β1 (transforming growth factor β1,TGF-β1)活性增高,引起骨转换加快、肌肉生成减少,出现骨骼病变和肌肉异常[2]。目前文献报道约250例PDD患者,TGFB1基因共检出12种突变类型[2-5]。
PDD好发于幼年,无性别及种族差异,疾病多呈进行性发展,也有自发缓解的报道[5]。该病呈高度表型异质性,主要表现为四肢骨痛、步态摇摆、肌肉萎缩、无力,也可出现第二性征发育不良,以及颅骨受累导致的面神经瘫痪、听力和视力异常等。典型的影像学表现为四肢长骨骨干皮质对称性、梭形增厚和硬化,髓腔变窄或消失,常不侵犯干骺端或骨骺,也可出现颅骨 (54% ~56.5%)、骨盆(63%)受累[2,5]。
目前国内对于PDD的临床表现型及致病基因突变型研究十分匮乏,不少患者未得到正确的诊断及治疗。本研究对一个罕见的PDD家系进行表型、基因型研究,并给予糖皮质激素治疗,前瞻性观察治疗对患者骨痛及活动障碍的影响,以深入了解该病的临床特征及发病机制。

表1 TGFB1基因PCR扩增引物序列
对象和方法
研究对象
患儿,女,7岁,主诉:骨痛、活动困难4年。于2015年3月就诊于北京协和医院。患者系第三胎第一产,母孕期平顺,足月剖宫产,出生体重3.2 kg,身长50 cm(体重及身长均处于第50百分位)。母乳喂养1年,坐起、萌牙时间与同龄儿无异,1岁会走路。3岁出现双下肢酸痛、无力,伴双膝关节伸展受限、步态摇摆,否认骨折史,无口干,无尿中排石。个人史:无特殊。家族史:父母非近亲婚配,否认类似患者。查体:身高118 cm,体重15.5 kg(分别位于同性别、同年龄儿童-1SD、-3SD),消瘦体型,步态摇摆,四肢肌肉萎缩;胸骨稍前突,双下肢呈X形畸形;双乳Ⅰ期,阴毛Ⅰ期。
本研究经北京协和医院伦理委员会批准。研究前征得患者及家属同意,签署知情同意书。
TGFB1基因检测
采集患者及父母外周静脉血,采用沉淀法提取白细胞基因组DNA(Tiangen试剂盒)。TGFB1基因共有7个外显子,采用Prime 3设计引物 (表1),扩增TGFB1的1~7所有编码外显子、外显子和内含子交界区及5'和3'侧翼序列。PCR反应体系共30 μl:Taq mix 15 μl、上下游引物各1 μl、基因组DNA 2 μl和ddH2O 11 μl。PCR反应条件为:预变性95℃,3 min;变性95℃,30 s;退火 58~63℃,30 s;延伸 72℃,1 min,循环35次;总延伸72℃,10 min。PCR扩增产物纯化后行Sanger测序,测序结果与参考序列NM_000660.5比对,以确定TGFB1突变。
血生化检查及骨骼影像学检查
分别在治疗前后,留取患者静脉血,分离血清后行常规生化及以下指标测定。使用罗氏电化学发光免疫分析仪测定血清 β-胶原降解产物 (β-isomerized carboxy-telopeptide of type I collagen,β-CTX,骨吸收指标,正常参考值0.21~0.44 ng/ml);血清总碱性磷酸酶 (alkaline phosphatase,ALP,骨形成指标,儿童参考值42~390 U/L);25羟维生素D(25 hydroxy vitamin D,25OHD,反映维生素 D营养状态,参考值50~125 nmol/L);全段甲状旁腺素 (intact parathyroid hormone,PTH,参考值12~65 ng/L)。完成头颅、胸腰椎、股骨、胫腓骨正侧位X线片,采用双能X线吸收仪 (GE Lunar DXA)测定第2~4腰椎后前位、股骨颈、大转子区及全髋骨密度 (g/cm2),参考同年龄、同性别正常儿童的参考范围计算骨密度Z值[6]。

图1 进行性骨干发育不良患者家系图 (A)及TGFB1基因测序结果 (B)
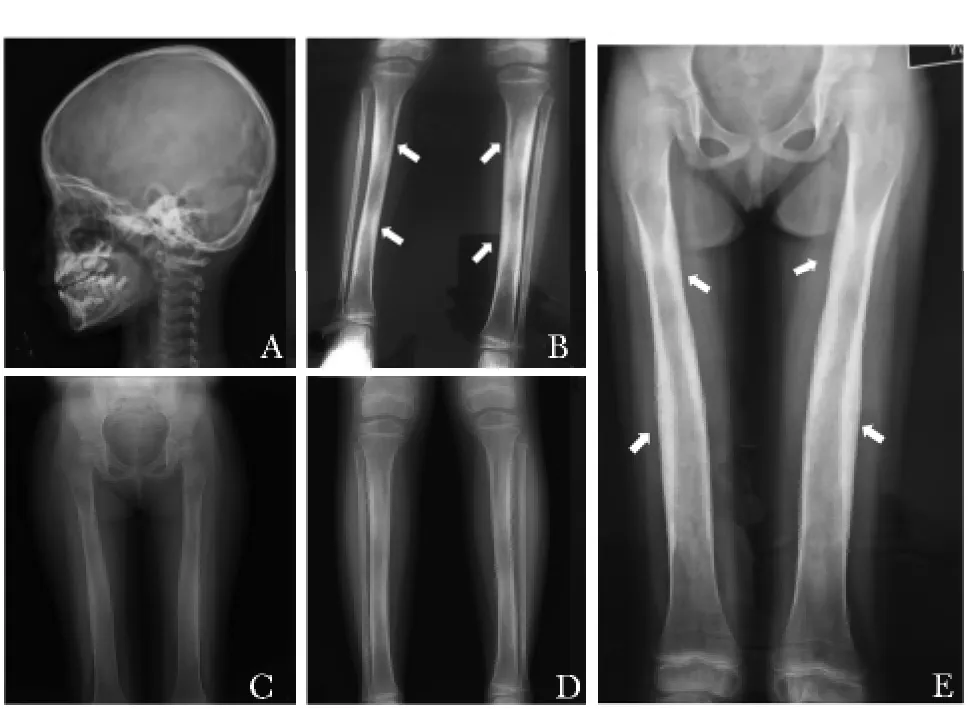
图2 进行性骨干发育不良患者的影像学表现
结果
TGFB1基因突变
患者TGFB1基因的第4外显子检出c.652C>T杂合性错义突变,导致编码的TGF-β1第218位氨基酸精氨酸被半胱氨酸替换 (p.Arg218Cys)。该突变位点为蛋白序列高度保守区,位于TGF-β1复合物的潜活相关肽 (latent associate peptide,LAP)上。患者父母的基因序列均未发现该突变,故此突变为一个新生突变 (de novo mutation)(图1)。本研究未发现TGFB1其他位点存在突变。
临床特点及治疗
患者生化检查示血钙、磷、ALP水平均位于年龄匹配的正常范围内,而血清β-CTX浓度较正常值升高3倍,提示存在骨转换加快;患者25OHD水平明显降低,提示维生素D缺乏,考虑与长期骨骼肌肉疼痛、户外活动减少有关;患者甲状腺功能正常,性激素为青春发育前正常水平。骨密度检查示髋部骨密度明显减低,提示骨质量下降、骨折风险增加 (表2)。四肢骨骼X线可见双股骨、胫腓骨、肱骨及尺桡骨骨干增粗,骨皮质不均匀增厚,骨小梁粗乱,相应骨髓腔变窄;颅骨未见异常 (图2)。
结合患者临床表现及基因异常,诊断为进行性骨干发育不良,予泼尼松20 mg/d起始治疗,4个月后逐渐减量至5 mg/d维持,同时每日补充钙剂500 mg、骨化三醇0.25 μg。经糖皮质激素治疗4个月,患者骨痛缓解,体重增加,步态恢复正常,活动能力明显改善;骨吸收指标β-CTX水平较治疗前下降30%,骨形成指标ALP下降7.5%(表2);骨骼X线片表现较前无加重 (图2)。泼尼松治疗过程中,患者血压、血糖、血常规、肝肾功、24 h尿钙均在正常范围内。
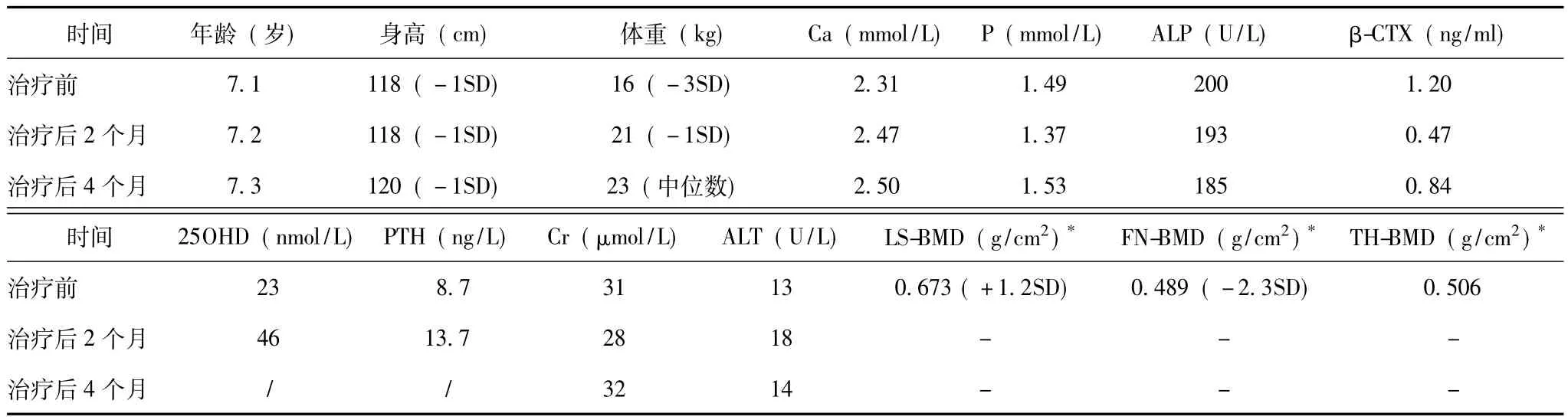
表2 进行性骨干发育不良患者治疗前后的临床特征
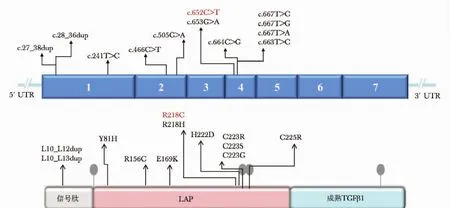
图3 进行性骨干发育不良TGFB1基因突变的DNA和蛋白位点
讨论
PDD是以四肢长骨骨干皮质梭形增厚为特点的罕见单基因遗传性骨骼疾病。该病由Cockayne于1920年首次描述,1922年由Camurati明确为遗传性骨病,1929年Engelmann首次报道PDD患者[3];直到2000年才将其致病基因定位于TGFB1[7]。本研究报道了1例典型的PDD儿童患者,表现为明显的进行性下肢骨痛、肌肉减少,影像学具有四肢骨皮质不均匀性增厚、硬化的特点,并经过基因突变检测,证实患者为TGFB1基因错义突变所致。
目前已报道的TGFB1基因突变有12种类型,主要位于第4外显子 (58%)和第1外显子 (25%),均位于TGF-β1复合物的信号肽和LAP区域,未发现TGF-β1区域突变 (图3)。所有突变可分为4种类型: (1)位于信号肽区的亮氨酸插入 (L10_L12dup、L10_ L13dup);(2)位于第1外显子、LAP的N末端错义突变 (Y81H);(3)位于第2外显子、LAP的N末端的错义突变 (R156C、E169K);(4)位于第4外显子、LAP区二硫键形成位点223Cys、225Cys附近的突变(218、222、223、225),其中218位点错义突变约占60%,为热点突变。本研究所发现的杂合、错义性突变 (c.652C>T,p.R218C)约占所有突变的40%,在中国人群已有报道[8],在此位点还有R218H的突变类型,均破坏了高度保守的疏水区、影响LAP二硫键形成,导致TGF-β1异常活化[9]。此外,PDD的疾病外显率有较大差异,有研究报道Y81H基因型的外显率较其他基因型低 (56%比99%),相关机制尚不清楚[2]。
目前TGFB1激活性突变引起PDD的机制尚不十分明确。TGFB1基因位于19q13,编码TGF-β1前体,包含氨基端信号肽、LAP和羧基端成熟TGF-β1;该前体经酶解后产生无活性的环形复合物,即二聚体LAP和二聚体TGF-β1,两者以非共价形式相连,从而阻止TGF-β1与受体结合及下游信号活化[10-11]。功能研究显示,位于TGFB1第4外显子的突变,即上述第4种突变类型,因靠近二硫键形成位点而影响LAP二聚体形成,LAP构象改变引起TGF-β1释放;而第1外显子的突变则可抑制TGF-β1的分泌,引起细胞内TGF-β1聚集、促进转录反应[9,12]。因此,上述所有PDD的致病基因突变均使得LAP和TGF-β1复合物的稳定性下降,循环和骨骼中的活性TGF-β1升高。活化的TGF-β1可促进骨形成和骨吸收,引起骨转换加快[10,13],本例患者骨转换生化指标升高、骨硬化表现与此一致。TGF-β1对骨骼的作用,一方面促进成骨细胞生成,另一方面可直接刺激破骨细胞生成,也可通过上调OPG、下调RANKL水平间接抑制破骨细胞活性[3]。研究显示,R218C基因型小鼠的TGF-β1活性升高,骨皮质增厚、骨强度下降、骨折率增加,免疫组化可见不同部位成骨细胞、破骨细胞聚集;而注射TGF-β受体拮抗剂后,上述异常均有显著改善[10]。此外,TGF-β1可抑制肌细胞成熟、干扰肌肉组织修复,也可抑制脂肪生成,这也导致PDD患者出现肌肉和皮下脂肪减少[2-3,14]。
PDD的诊断需结合临床症状、影像学表现和基因异常。PDD患者常见临床表现有四肢骨痛 (68%)、步态异常 (48%)、疲乏 (44%)、肌肉减少 (39%)、脂肪减少 (21%)、听力下降 (15%)[2]。骨骼异常表现为骨质硬化、骨质疏松、骨软化,可出现关节屈曲挛缩,严重者有四肢细长、前额突出等类马凡体征[3]。PDD也可合并青春发育延迟、性腺功能减低,考虑与慢性疼痛、低体重指数、脂肪组织分泌脂肪因子减少有关,TGFB1突变也可能参与其中[15]。94%的PDD患者具有典型的骨骼X线表现,即长骨骨干对称性、不均匀性增厚,可进行性发展至干骺端,但无骨骺受累;骨显像则有助于疾病的早期诊断[2]。本研究中PDD患者表型与既往报道相一致,骨骼表现为骨量增加和骨量减少并存、骨折风险增加,目前尚未观察到青春发育迹象;因此除随访骨骼异常外,还应监测青春发育情况、骨龄、性激素水平。基于该病为常染色体显性遗传,明确基因诊断对该患者产前咨询有重要意义。然而PDD缺乏明确的基因型-表型间的关联,同一家族、相同基因型携带者的疾病外显率、临床表型也呈高度异质性,其潜在遗传学机制尚不清楚,可能受其他遗传因素、修饰基因的影响[2-3]。
PDD治疗方面,可能有效的药物包括糖皮质激素、双膦酸盐、降钙素、非甾体类抗炎药及手术治疗。糖皮质激素因抑制骨形成、降低骨密度,可有效缓解骨痛、改善影像学异常;通常泼尼松起始剂量1~2 mg/(kg·d),快速减量至最小有效剂量以维持治疗[2]。但长期糖皮质激素治疗可能导致药物性库欣综合征,出现高血压、高血糖、骨量丢失、骨折风险增加,影响儿童线性生长等不良反应。本研究患者接受泼尼松治疗4个月后,有效降低了骨转换生化指标、减轻了患者骨痛,提高了活动能力,短期应用尚未观察到明显不良反应,但未来患者长期治疗方案,需权衡糖皮质激素使用的利弊,慎重制定更为合理的治疗方案。目前有个案报道阿仑膦酸钠、帕米膦酸钠、唑来膦酸钠及降钙素通过抑制骨吸收而缓解PDD病情[16-18],但也有报道双膦酸盐可加重骨痛[19],故其应用于PDD的治疗尚存争议。此外,近期研究显示血管紧张素Ⅱ受体拮抗剂可抑制TGF-β1信号通路活性,氯沙坦0.75 mg/(kg·d)起始治疗不仅能缓解骨痛、提高运动耐量,也可有效地提高肌肉和脂肪含量、改善身体成分[20-21],这为PDD治疗提供了新思路,但其长期治疗的有效性和安全性仍有待大样本的前瞻性研究进一步证实。
综上,PDD是一种罕见的常染色体显性遗传性骨病,本研究证实TGFB1第218位错义突变是其可能的致病基因,TGFB1激活性突变导致的四肢骨痛、肌肉减少和骨干皮质增厚是疾病的典型临床表现。糖皮质激素治疗可以明显改善PDD患者的临床症状。本研究结果有助于提高对罕见疾病PDD的认识、分子诊断及治疗水平,但其潜在致病机制仍需深入研究。
[1]Bhadada SK,Sridhar S,Steenackers E,et al.Camurati-Engelmann disease(progressive diaphyseal dysplasia):reports of an Indian kindred[J].Calcif Tissue Int,2014,94:240-247.
[2]Janssens K,Vanhoenacker F,Bonduelle M,et al.Camurati-Engelmann disease:review of the clinical,radiological,and molecular data of 24 families and implications for diagnosis and treatment[J].J Med Genet,2006,43:1-11.
[3]Whyte MP,Totty WG,Novack DV,et al.Camurati-Engelmann disease:unique variant featuring a novel mutation in TGF-beta1 encoding transforming growth factor beta 1 and a missense change in TNFSF11 encoding RANK ligand[J].J Bone Miner Res,2011,26:920-933.
[4]Wu S,Liang S,Yan Y,et al.A novel mutation of TGF-beta1 in a Chinese family with Camurati-Engelmann disease[J].Bone,2007,40:1630-1634.
[5]Carlson ML,Beatty CW,Neff BA,et al.Skull base manifestations of Camurati-Engelmann disease[J].Arch Otolaryngol Head Neck Surg,2010,136:566-575.
[6]Khadilkar AV,Sanwalka NJ,Chiplonkar SA,et al.Normative data and percentile curves for Dual Energy X-ray Absorptiometry in healthy Indian girls and boys aged 5-17 years[J].Bone,2011,48:810-819.
[7]Kinoshita A,Saito T,Tomita H,et al.Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease[J].Nat Genet,2000,26:19-20.
[8]Wang C,Zhang BH,Liu YJ,et al.Transforming growth factor-beta1 gene mutations and phenotypes in pediatric patients with Camurati-Engelmann disease[J].Mol Med Rep,2013,7:1695-1699.
[9]Walton KL,Makanji Y,Chen J,et al.Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-beta1 complex[J].J Biol Chem,2010,285:17029-17037.
[10]Tang Y,Wu X,Lei W,et al.TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation[J].Nat Med,2009,15:757-765.
[11]Shi M,Zhu J,Wang R,et al.Latent TGF-beta structure and activation[J].Nature,2011,474:343-349.
[12]Janssens K,Ten DP,Ralston SH,et al.Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein[J].J Biol Chem,2003,278:7718-7724.
[13]Chen G,Deng C,Li YP.TGF-beta and BMP signaling in osteoblast differentiation and bone formation[J].Int J Biol Sci,2012,8:272-288.
[14]Zamani N,Brown CW.Emerging roles for the transforming growth factor-beta superfamily in regulating adiposity and energy expenditure[J].Endocr Rev,2011,32:387-403.
[15]Toumba M,Neocleous V,Shammas C,et al.A family with Camurati-Engelman disease:the role of the missense p.R218C mutation in TGFbeta1 in bones and endocrine glands[J].J Pediatr Endocrinol Metab,2013,26:1189-1195.
[16]Iba K,Takada J,Kamasaki H,et al.A significant improvement in lower limb pain after treatment with alendronate in two cases of Camurati-Engelmann disease[J].J Bone Miner Metab,2008,26:107-109.
[17]Savoie A,Gouin F,Maugars Y,et al.Treatment responses in five patients with Ribbing disease including two with 466 C>T missense mutations in TGFbeta1[J].Joint Bone Spine,2013,80:638-644.
[18]Trombetti A,Cortes F,Kaelin A,et al.Intranasal calcitonin reducing bone pain in a patient with Camurati-Engelmann disease[J].Scand J Rheumatol,2012,41:75-77.
[19]Castro GR,Appenzeller S,Marques-Neto JF,et al.Camurati-Engelmann disease:failure of response to bisphosphonates:report of two cases[J].Clin Rheumatol,2005,24:398-401.
[20]Ayyavoo A,Derraik JG,Cutfield WS,et al.Elimination of pain and improvement of exercise capacity in Camurati-Engelmann disease with losartan[J].J Clin Endocrinol Metab,2014,99:3978-3982.
[21]Simsek-Kiper PO,Dikoglu E,Campos-Xavier B,et al.Positive effects of an angiotensin II type 1 receptor antagonist in Camurati-Engelmann disease:a single case observation[J].Am J Med Genet A,2014,164A:2667-2671.
Mutation in TGFB1 Causes Rare Progressive Diaphyseal Dysplasia
XU Xiao-jie,MA Dou-dou,LFang,LIU Yi,WANG Jian-yi,JIANG Yan,WANG Ou,XIA Wei-bo,XING Xiao-ping,LI Mei
Department of Endocrinology,Key Laboratory of Endocrinology of Ministry of Health,Peking Union Medical College Hospital,Chinese Academy of Medical Sciences&Peking Union Medical College,Beijing 100730,China
ObjectiveTo investigate the phenotypes of a kindred with progressive diaphyseal dysplasia (PDD)and to detect the mutation of transforming growth factor beta-1(TGFB1)gene.MethodsA PDD patient of a non-consanguineous family presented with early onset in childhood,who suffered from lower limb pain,fatigability and muscle weakness.Her clinical manifestations,features of skeletal X-ray examination,and bone turnover markers were evaluated.Mutation of TGFB1 was identified by direct Sanger sequencing of polymerase chain reaction amplification product.ResultsThe proband presented with elevated bone turnover biomarkers,and nonuniform thickening and sclerosis of bone cortex of limbs in X-ray films.A heterozygous missense mutation c.652C>T(p.Arg218Cys)in exon 4 of TGFB1 was identified in the proband,but not in either of her parents.Glucocorticoid was given and after 4 months of treatment,the bone pain and activity were obviously improved.ConclusionsThe typical clinical manifestations of PDD are limb pain and diaphyseal hyperostosis.The missense mutations at position 218 of TGFB1 are hotspot pathogenic mutations of PDD.Glucocorticoids can mitigate the symptoms in PDD patients.
progressive diaphyseal dysplasia;phenotype;TGFB1 gene;mutation
LI Mei Tel:010-69155088,E-mail:limeilzh@sina.com
R681
A
1674-9081(2015)05-0327-06
10.3969/j.issn.1674-9081.2015.05.003
2015-07-18)
李 梅 电话:010-69155088,E-mail:limeilzh@sina.com
国家自然科学基金面上项目 (8100623)及国家临床重点专科建设项目 (WBYZ2011-873)
