下调microRNA-181b在小鼠缺血性脑损伤中的神经保护作用*
2015-05-16彭志锋
彭志锋
(山西大同大学医学院生理教研室,山西 大同 037009)
下调microRNA-181b在小鼠缺血性脑损伤中的神经保护作用*
彭志锋△
(山西大同大学医学院生理教研室,山西 大同 037009)
目的:探讨microRNA-181b(miR-181b)在缺血性小鼠脑损伤病理过程中的神经保护作用。方法:应用N2A细胞氧-糖剥夺(OGD)模型模拟神经细胞缺血损伤;应用小鼠大脑中动脉阻塞(MCAO)模型模拟缺血性脑损伤(成功率约60%),原位细胞凋亡检测试剂盒检测N2A细胞损伤程度,蛋白印迹检测miR-181b靶蛋白自噬蛋白5(Atg5)及caspase-9的变化情况,萤光素酶报告基因技术检测miR-181b对Atg5 mRNA的直接调控作用。结果:在OGD致N2A细胞缺血损伤过程中,通过上调或抑制miR-181b的表达水平可以显著影响N2A细胞的凋亡(P<0.05);miR-181b表达水平的改变可显著影响Atg5蛋白表达水平(P<0.05);共转染miR-181b前体或miR-181b抑制剂可显著抑制或增高含有Atg5 mRNA 3’-UTR的萤光素酶报告基因的活性(P<0.05);MCAO后活化caspase-9的表达显著升高,而miR-181b拮抗剂可使MCAO后剪切的caspase-9表达明显减少(P<0.05)。结论:下调miR-181b可能通过调节Atg5的蛋白表达在缺血性小鼠脑损伤中发挥神经保护作用。
大脑中动脉阻塞;氧-糖剥夺;缺血性损伤;微小RNA-181b;自噬蛋白5;N2A细胞
脑卒中是当今世界严重危害人类健康和生命安全的难治性疾病,具有发病率高、致残率高和死亡率高的特征。我国也是脑卒中高发大国,每年新发病200万人,死亡约150万人,是继肿瘤之后导致城乡居民死亡的第二杀手[1]。短暂或持久的局灶性脑缺血可引起一系列病理生理变化而导致脑损伤。由此引发的能量代谢障碍、兴奋性氨基酸毒性作用、炎症反应、半暗带去极化及细胞凋亡等共同参与了其病理生理变化过程[2]。因其发病机制比较复杂,目前临床上对脑卒中的治疗尚缺乏有效措施。
大量的研究证实,丝裂原活化蛋白激酶、PI3K/ Akt、PKA、JAK-STAT、Notch、Toll样受体等信号转导通路参与了缺血性脑卒中的发生、发展过程[3-8]。然而这些信号通路不能完全阐明缺血性脑卒中的发病机制。微小RNA(microRNAs,miRANs)作为内源性的、非编码的小RNA分子,至少调节细胞中30%的基因,从而引起了许多研究者的关注。近年来,越来越多的证据显示,miRNAs在缺血性脑损伤中起着重要的作用。应用miRNA高通量方法检测大脑中动脉阻塞(middle cerebral artery occlusion,MCAO)模型缺血再灌注后大鼠血液和脑内的miRNA情况,鉴定出7类miRNAs,其中有些miRNAs的调节在脑缺血进程中发挥重要作用[9]。短暂的MCAO后再灌注不同时点成年大鼠脑内miRNAs出现不同的表达情况[10]。
最近,文献报道[11]通过miRNA基因芯片技术,在MCAO小鼠脑皮层筛选出差异性表达的microRNA-181b(miR-181b)。通过生物信息学分析,miR-181b有可能靶向调节自噬蛋白5(autophagy protein 5,Atg5)。然而,关于miR-181b在缺血性小鼠脑损伤中的作用,以及其是否通过调节Atg5蛋白表达水平发挥作用尚未见报道。因此,本文利用N2A细胞的氧-糖剥夺(oxygen-glucose depletion,OGD)模型和MCAO致小鼠脑缺血模型,探讨miR-181b在缺血性小鼠脑损伤中作用及其对Atg5蛋白表达的影响。
材料和方法
1 动物
健康成年、雄性BALB/c小鼠,体重18~22 g,购自首都医科大学动物部,许可证号为SCXK-(军) 2007-004。
2 主要试剂和仪器
胎牛血清、DMEM、OPTI-MEM I培养基(Gibco); Lipofectamine 2000(Invitrogen);Pre-miR-181b、AntimiR-181b(Ambion);微量注射泵(DURECT);miR-181b拮抗剂(Ribobio);兔抗Atg5多克隆抗体、鼠抗β-actin多克隆抗体(Sigma);兔抗caspase-9、cleavedcaspase-9多克隆抗体(CST);萤光检测试剂盒(Promega);原位细胞凋亡POD检测试剂盒(Roche)。
3 主要方法
3.1 大脑中动脉阻塞诱导局部脑缺血再灌模型的制备腹腔注射1%戊巴比妥钠(0.06 g/kg)麻醉小鼠,自颈部行1 cm长的正中纵切口,钝性分离下颌下腺,暴露胸锁乳突肌。手术显微镜下暴露左侧颈动脉鞘并游离出颈总动脉、颈外动脉及颈内动脉,小心保护与之伴行的迷走神经,然后用5/0缝合丝线双重结扎颈总动脉近心端和颈外动脉远心端;在颈总动脉上两线结间打一松结并轻轻提起以阻断血流,在颈外动脉远端的动脉壁上用针头扎1个小孔,将制备好的线栓从小孔插入颈总动脉至颈内动脉并进而向上到达大脑中动脉直至遇到轻微阻力(深度约12.0 mm),固定线栓,将下颌下腺归位,20 min后拔出线栓,缝合皮肤。10只小鼠做MCAO手术,死亡4只小鼠,成功率约60%。假手术组(sham)只进行手术操作,不插入线栓。
3.2 脑室给药小鼠在缺血手术前先经1%戊巴比妥钠腹腔注射麻醉后固定于小鼠脑立体定位仪上,局部用利多卡因局麻后行正中切口,按Paxinos的侧脑室注射方法,在立体定位仪的引导下将脑室注射套管插入到左则脑室,然后用胶水固定,缝合伤口。接着把微量注射泵埋植在腹腔内,通过导管把微量注射泵和套管连接起来。术后腹腔注射青霉素钠8×105U/d,预防感染。在小鼠MCAO前2 d持续将miR-181b拮抗剂、阴性对照(5 μmol/L,1 μL/h)注入。在微量注射泵移植前1 d,miR-181b拮抗剂及阴性对照填充到微量注射泵中37°C过夜。MCAO后24 h取出脑组织进行相应的分析。
3.3 OGD模型制备在5%CO2、95%O2、37°C的培养箱中,用含有10%FBS的DMEM完全培养基对N2A细胞进行培养[10]。根据实验要求进行细胞传代,在保证细胞达到实验密度并处于生长对数期的状态下,按不同实验要求处理细胞。在离体细胞水平模拟缺血/再灌注刺激,采用OGD细胞模型。将N2A细胞接种于含完全培养基(DMEM+10% FBS)的96孔板中,置37℃、5%CO2、95%O2的培养箱内培养。次日将完全培养基更换为无糖DMEM培养基,并通以混合低氧气体(5%CO2、1%O2和94%N2)。完成3 h OGD刺激后,将无糖DMEM培养基更换为含10%FBS的DMEM高糖培养基(复糖)同时,于5%CO2、95%O2(复氧)、37℃的条件下继续培养24 h后,离体细胞水平观察缺血/再灌对N2A细胞的损伤情况。
3.4转染N2A细胞转染前1 d,胰酶消化细胞并计数后,将N2A细胞接种于96孔板内,使其在转染时密度为50%。次日使用Lipofectamine 2 000分别转染pre-miR-181b、anti-miR-181b及相应pre-miR对照、anti-miR对照。具体方法如下:对于每孔细胞,分别使用50 μL OPTI-MEM I培养基稀释miRNA核苷酸片段和Lipofectamine 2 000转染试剂,并静置Lipofectamine 2 000稀释液5 min。混合稀释后的miRNA核苷酸片段和Lipofectamine 2 000,在室温静置20 min。用PBS轻轻冲洗培养孔,镜下观察部分细胞略变圆,但没有细胞悬浮。吸去PBS,缓慢加入上述混合物,摇动培养板,轻轻混匀。将细胞置于37℃、5%CO2、95%O2培养箱中继续培养;6 h后将转染液更换为完全培养基,48 h后收集细胞或进行OGD处理,通过实时定量PCR验证转染是否成功。
3.5 蛋白印迹实验取30 μg进行SDS-PAGE分离蛋白,后转至PVDF膜上。经5%牛奶封闭1 h后,取出并用含Tween-20的磷酸盐缓冲液(PBST)洗膜3次,每次10 min。用兔抗Atg5多克隆抗体(1∶1 000)和鼠抗β-actin多克隆抗体(1∶2 000)4℃孵育3 h,PBST洗3次,每次10 min。山羊抗兔和山羊抗鼠II抗(1∶25 000),室温孵育1 h。PBST洗3次,每次5 min,后用化学发光扫描系统检测并摄片,以β-actin为内参照,应用图像分析软件Quantity one v4.62进行吸光度积分值分析。
3.6 原位细胞凋亡检测经3 h OGD/24 h复糖复氧刺激诱导细胞凋亡;4%多聚甲醛中固定细胞1 h; PBS浸洗2次,每次5 min;蛋白激酶K(5 Mg/L)5 min;PBST 10 min;PBS浸洗2次,每次5 min;用滤纸吸去样本区域周围的多余液体,滴加50 μL的TdT酶反应液于样本区域上,于37℃、潮湿环境中反应60 min;PBS浸洗3次,每次5 min;POD 1 h;PBS浸洗3次,每次5 min;于载玻片的样本区域滴加100 μL DAB工作液(直到呈现浅棕色背景);用去离子水漂洗2~3次;用滤纸小心吸去载玻片上的多余液体,滴加3滴甘油于样本区域,然后盖上盖玻片进行封片,即可在光学显微镜下进行观察并拍照。
3.7 双萤光素酶报告基因检测含有Atg5 mRNA 3’-UTR或突变3’-UTR(Mut)萤光素酶报告基因由广州市锐博生物科技有限公司构建,将该嵌合载体与Pre-miR-181b或Anti-miR-181b等共同转染N2A细胞,接种密度为(5~6)×107/L。5%CO2、95% O2、37℃孵育48 h后,终止培养。应用Promega的萤光检测试剂盒进行萤光强度测定。用萤火虫萤光素酶为内参照作比值,从而得出相对的萤光强度比,并将数据进行分析。
4 统计学处理
实验数据用SPSS 11.5统计软件进行单因素方差分析(one-way ANOVA),计量资料以均数±标准误(mean±SEM)表示,以P<0.05为差异有统计学意义。
结果
1 miR-181b对OGD致N2A细胞凋亡的影响
原位细胞凋亡检测结果显示,pre-miR-181b可明显增加OGD致N2A细胞的凋亡,而anti-miR-181b可使OGD处理的N2A细胞凋亡减弱(P<0.05),见图1。
2 miR-181b调节Atg5蛋白的表达水平
蛋白印迹检测结果表明,转染pre-miR-181b过表达miR-181b时,Atg5蛋白表达量显著降低;而转染anti-miR-181抑制miR-181b表达后,Atg5蛋白表达水平明显升高(P<0.05),见图2。

Figure 2.The effects of miR-181b on Atg5 protein levels in N2A cells determined by Western blotting.A:vehicle;B: anti-miR-181b;C:anti-miR control;D:pre-miR-181b;E:pre-miR control.Mean±SEM.n=3.*P<0.05 vs A.图2 miR-181b对N2A细胞Atg5蛋白的调节
3 miR-181b与Atg5 mRNA的3’-UTR相结合负性调节其翻译
萤光素酶报告基因检测结果示,pre-miR-181b显著抑制萤光素酶活性,而转染anti-miR-181b则萤光强度明显升高(P<0.05),见图3。结果表明miR-181b直接与Atg5 mRNA的3’-UTR结合,从而负性调控Atg5的蛋白表达水平。
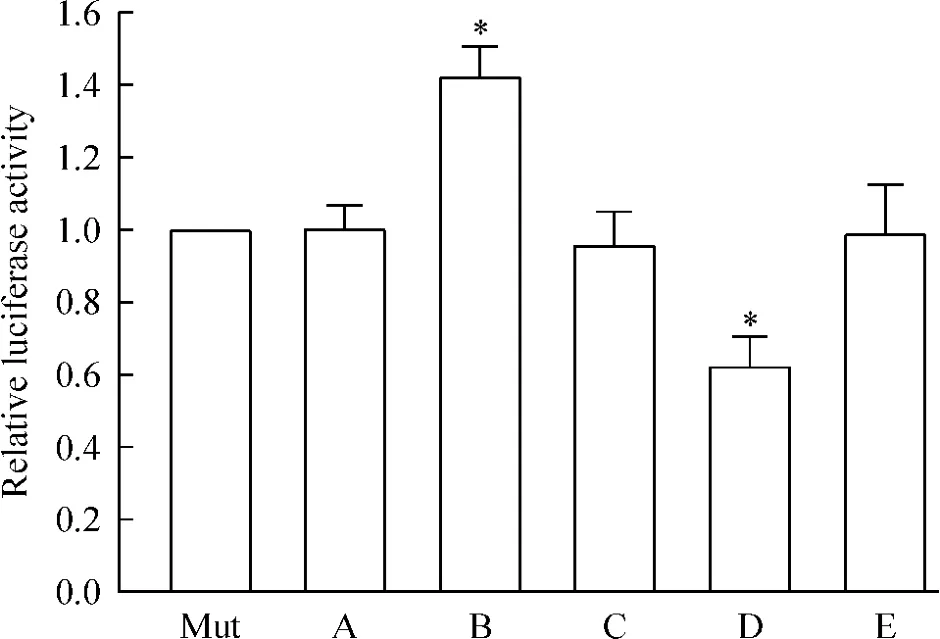
Figure 3.The effect of miR-181b on the luciferase activities of luciferase expressing plasmid containing 3’-UTR of Atg5 mRNA.A:vehicle;B:pre-miR-181b;C:premiR control;D:anti-miR-181b;E:anti-miR control.Mean±SEM.n=5.*P<0.05 vs Mut.图3 miR-181b对含Atg5 mRNA的3’-UTR萤光素酶表达的影响
4 miR-181b拮抗剂对活化的caspase-9的影响
MCAO后激活的caspase-9与非手术组相比显著提高;miR-181b拮抗剂可以使MCAO后deaved caspase-9明显减少(P<0.05),见图4。结果提示miR-181b在小鼠缺血性损伤中发挥作用,抑制miR-181b可以缓解小鼠脑缺血引起的凋亡。

Figure 4.The protein levels of cleaved caspase-9 after MCAO in mice with miR-181b antagonist.A:sham;B:MCAO; C:MCAO+antagonist control;D:MCAO+miR-181b antagonist.Mean±SEM.n=3.*P<0.05 vs A;#P<0.05 vs C.图4 miR-181b拮抗剂对活化caspase-9的影响
讨论
本研究证实过表达miR-181b可使OGD引起的N2A细胞死亡增加,而降低miR-181b表达水平则可减轻OGD致N2A细胞的缺血损伤,其作用机制可能与负性调节Atg5蛋白表达水平相关;抑制miR-181b可减轻MCAO诱导小鼠脑神经元的凋亡。
缺血性脑卒中后神经细胞损伤主要表现为坏死和凋亡。当脑缺血发生时,缺血中心区的神经细胞由于血流供应的急速缺乏而迅速发生坏死性死亡;但在中心缺血区外围,一些细胞会经过一段时间后慢慢地死亡,这些细胞的死亡常常呈现凋亡特征,这种脑缺血后半影区的神经细胞缺失即为凋亡,而凋亡引起的损伤更严重、更持久,且在发病机制中占主导地位[12]。因此,采取积极有效措施,阻止缺血半影区神经细胞的凋亡,已成为当前研究的热点。本实验首次提出了下调miR-181b可能是通过上调其靶基因Atg发挥神经保护作用。Atg5是自噬相关基因,敲除Atg5可导致小鼠自噬功能不全而于出生后数小时内死亡[13]。自噬是真核生物进化上高度保守的分级代谢方式,通过溶酶体降解细胞内受损的细胞器及大分子物质来维护细胞代谢平衡和细胞内环境稳定,促进细胞生长、增殖[14]。陆续有研究在局灶性脑缺血、全脑缺血及低氧-缺血模型中发现了自噬现象[15]。在大鼠脑缺血模型研究中观察到,缺血灶周围脑组织神经元中的溶酶体和自噬活动均显著增强,认为自噬活动对缺血周围脑组织具有保护作用[16]。在低氧/缺血模型中,给予自噬诱导剂雷帕霉素能够减少低氧/缺血损伤[17],提示了自噬具有神经保护作用[18],该过程的自噬可观察到显著的脑保护现象。另外还有研究报道自噬在缺血/复灌模型中通过抑制细胞凋亡发挥了神经保护作用[19]。自噬在缺血性脑损伤过程中发挥保护作用的机制尚不完全明确。相关研究提示自噬通过清除损伤线粒体,即线粒体自噬,可能在其中发挥了关键作用。损伤的线粒体在缺血性脑损伤过程中释放包括细胞色素C在内的多种蛋白,进而诱发线粒体依赖的神经元凋亡[20]。
[1]张婷婷,谢谦,刘腾远,等.MicroRNAs与脑卒中的研究进展[J].分子诊断与治疗杂志,2012,4(1):1-10.
[2]Moskowitz MA,Lo EH,Iadecola C.The science of stroke:mechanisms in search of treatments[J].Neuron,2010,67(2):181-198.
[3]Bohacek I,Cordeau P,Lalancette-Hebert M,et al.Tolllike receptor 2 deficiency leads to delayed exacerbation of ischemic injury[J].J Neuroinflammation,2012,9:191.
[4]Hara T,Hamada J,Yano S,et al.CREB is required for acquisition of ischemic tolerance in gerbil hippocampal CA1 region[J].J Neurochem,2003,86(4):805-814.
[5]Justicia C,Gabriel C,Planas AM.Activation of the JAK/ STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes[J].Glia,2000,30(3):253-270.
[6]Noshita N,Lewen A,Sugawara T,et al.Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice[J].J Cereb Blood Flow Metab,2001,21(12):1442-1450.
[7]Arumugam TV,Chan SL,Jo DG,et al.Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke[J].Nat Med,2006,12(6):621-623.
[8]Wang X,Wang H,Xu L,et al.Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice:exploration of potential mechanism associated with apoptosis[J].J Pharmacol Exp Ther,2003,304(1):172-178.
[9]Jeyaseelan K,Lim KY,Armugam A.MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion[J].Stroke,2008,39(3):959-966.
[10]Dharap A,Bowen K,Place R,et al.Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome[J].J Cereb Blood Flow Metab,2009,29 (4):675-687.
[11]Liu C,Peng Z,Zhang N,et al.Identification of differentially expressed microRNAs and their PKC-isoform specific gene network prediction during hypoxic pre-conditioning and focal cerebral ischemia of mice[J].J Neurochem,2012,120(5):830-841.
[12]Rami A,Kogel D.Apoptosis meets autophagy-like cell death in the ischemic penumbra:Two sides of the same coin?[J].Autophagy,2008,4(4):422-426.
[13]Kuma A,Hatano M,Matsui M,et al.The role of autophagy during the early neonatal starvation period[J].Nature,2004,432(7020):1032-1036.
[14]Zou M,Lu N,Hu C,et al.Beclin 1-mediated autophagy in hepatocellular carcinoma cells:implication in anticancer efficiency of oroxylin A via inhibition of mTOR signaling[J].Cell Signal,2012,24(8):1722-1732.
[15]Wen YD,Sheng R,Zhang LS,et al.Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways[J].Autophagy,2008,4(6):762-769.
[16]Puyal J,Clarke PG.Targeting autophagy to prevent neonatal stroke damage[J].Autophagy,2009,5(7): 1060-1061.
[17]Carloni S,Girelli S,Scopa C,et al.Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia[J].Autophagy,2010,6(3):366-377.
[18]Koike M,Shibata M,Tadakoshi M,et al.Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury[J].Am J Pathol,2008,172(2):454-469.
[19]Zhang X,Yan H,Yuan Y,et al.Cerebral ischemiareperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance[J].Autophagy,2013,9(9):1321-1333.
[20]Jemmerson R,Dubinsky JM,Brustovetsky N.Cytochrome C release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases[J].Antioxid Redox Signal,2005,7(9-10):1158-1172.
Down-regulation of microRNA-181b has protective effect on cerebral ischemic injury of mice
PENG Zhi-feng
(Department of Physiology,School of Medicine,Shanxi Datong University,Datong 037009,China.E-mail:pzf181@126.com)
AIM:To explore the role of microRNA-181b(miR-181b)in ischemic injury and autophagy protein 5(Atg5)levels of mice.METHODS:Oxygen-glucose depletion(OGD)model in N2A cells to mimic ischemic injury in vitro was established.A middle cerebral artery occlusion(MCAO)model to mimic ischemic injury in vivo was also induced in mice.The N2A cell apoptosis after OGD was assessed by in situ cell death detection kit.The Atg5 and caspase-9 expressions were determined by Western blotting.Luciferase reporter assay was performed to identify the direct binding of miR-181b with 3’-UTR of Atg5 mRNA.RESULTS:The alteration of miR-181b expression level by transfection with premiR-181b or anti-miR-181b significantly affected N2A cell apoptosis(P<0.05).Accordingly,the changes of miR-181b levels significantly altered the protein level of Atg5(P<0.05).Co-transfection of the luciferase reporters with pre-miR-181b or anti-miR-181b resulted in the inhibition or enhancement of the luciferase activities of luciferase expressing plasmid containing 3’-UTR of Atg5 mRNA(P<0.05).In addition,the miR-181b antagonist significantly reduced the cleaved caspase-9 levels in cerebral ischemic cortex of the mice after MCAO(P<0.05).CONCLUSION:Down-regulation of miR-181b plays an important role in ischemic injury of mice through regulating Atg5 protein level.
Middle cerebral artery occlusion;Oxygen-glucose depletion;Ischemic injury;MicroRNA-181b; Autophagy protein 5;N2A cells
R363;R338
A
10.3969/j.issn.1000-4718.2015.02.006
1000-4718(2015)02-224-05
2014-08-25
2014-11-18
山西大同大学博士科研启动经费
△通讯作者Tel:0352-6205665;E-mail:pzf181@126.com
