铑催化线性氢甲酰化反应的研究进展
2015-05-14陈才友张绪穆
陈才友,吕 辉,张绪穆
(武汉大学 化学与分子科学学院,湖北 武汉 430072)
氢甲酰化反应又称羰基合成反应,是一种利用过渡金属催化剂将烯烃、氢气、一氧化碳转化为醛类化合物的反应。由于该反应可将石油工业中常见的烯烃转化为醛并实现碳链增长,因而在工业上有重要的应用。
氢甲酰化反应通常可生成线性醛或支链醛,其中,线性醛在工业中被广泛用于合成洗涤剂和增塑剂;支链醛则被广泛用于药物及化学试剂的合成。此外,醛类化合物还是一种重要的化工原料或反应中间体,可进一步通过化学反应(如氢化、氧化、还原氨化等)生成醇、羧酸、胺、酯等极具价值的化学物质[1-4]。氢甲酰化反应生成的醛可应用于合成吲哚(Fischer吲哚合成法)。基于醛类化合物具有重要用途,氢甲酰化反应在工业上已得到广泛应用,且已成为现代工业中最重要的均相催化反应之一。目前,工业上每年通过氢甲酰化反应生产的醛类物质已达到104kt[4]。
氢甲酰化反应在生成线性氢甲酰化产物和支链氢甲酰化产物的同时,还会发生其他类型的反应,如烯烃类底物异构化生成相应的异构化烯烃,或通过氢化反应生成相应的烷烃等。因此,提高氢甲酰化反应的选择性、高收率获得目标产物是需要解决的重要问题,而解决这一问题最重要的方法是开发高效的氢甲酰化配体。
早期Rh催化线性氢甲酰化反应使用的是单齿膦配体,由于单齿膦配体的配位效应不强,为获得高选择性,反应中需加入大大过量的单齿膦配体[5],但大大过量的配体不利于后续的分离操作,同时也不经济、不环保。为此,研究者开发了双齿膦配体。由于双齿膦配体与Rh的配位作用远强于单齿膦配体,双齿膦配体在线性氢甲酰化反应中具有比单齿膦配体更高的选择性。随后,在双齿膦配体的基础上,研究者设计并合成了四齿膦配体。四齿膦配体在线性氢甲酰化反应中的选择性又远强于双齿膦配体,如在烯烃、烯丙基酯、烯丙基腈类化合物的线性氢甲酰化反应中,四齿膦配体的选择性均高于双齿膦配体。最近开发的新型三齿膦配体在烯烃的线性氢甲酰化反应中也表现出非常高的活性和选择性。
本文将以氢甲酰化的反应机理以及线性氢甲酰化反应中配体的发展和应用为主线,对Rh催化的线性氢甲酰化反应进行介绍。
1 氢甲酰化反应机理
氢甲酰化反应最初由Roelen教授于1938年发现[5],随后在工业中广泛应用,并已成为现代工业中最大规模的均相催化反应之一。在氢甲酰化反应过程中生成线性羰基化产物的过程被称为线性氢甲酰化反应。Rh催化的氢甲酰化反应主要按图1所示进行[6]。
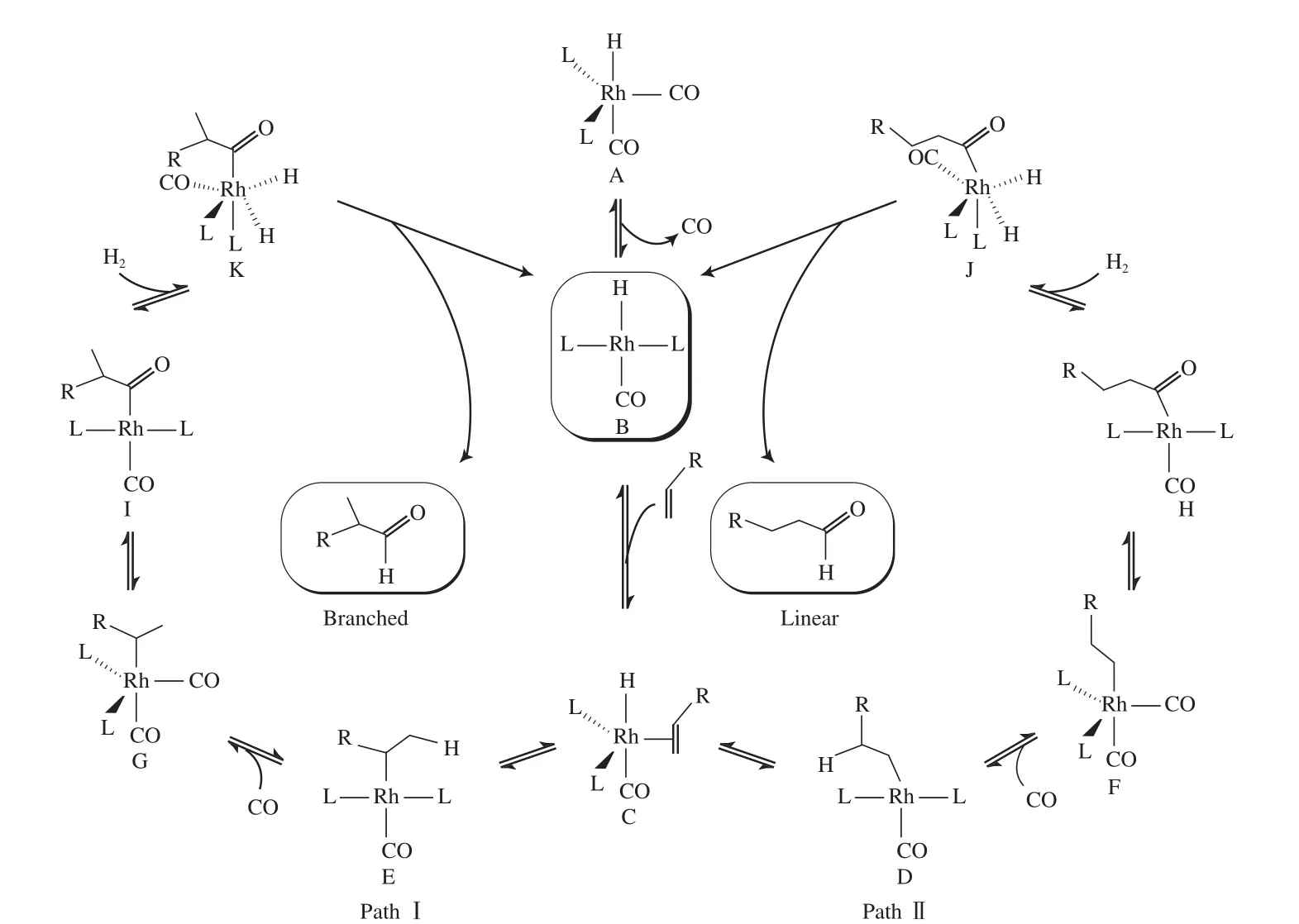
图1 Rh催化的氢甲酰化反应Fig.1 Paths for Rh catalyzed hydroformylation.
从图1可看出,首先,Rh催化剂(A)离去一分子CO形成带一个空配位点的催化形态(B),随后烯烃配位到Rh中心上,形成中间体(C)。此时反应有两种可能的反应途径:1)按PathⅠ途径进行,中间体(C)经烯烃插入生成烯烃β碳原子直接键连到Rh中心的中间体(E);随后CO插入并与Rh中心配位形成中间体(I);接着发生H2对中间体的氧化加成,再经还原消除反应生成支链氢甲酰化产物,同时Rh催化剂还原成原始的催化形态(B),从而完成一个催化循环过程。2)按PathⅡ途径进行,基本过程与PathⅠ类似,但在中间体(C)发生烯烃插入时,烯烃的端位α碳原子直接键连到Rh中心上形成中间体(D),然后按PathⅠ所示类似过程,生成线性氢甲酰化产物。
2 线性氢甲酰化配体的发展
20世纪60年代末,Evans等[7]首次用Rh络合物HRh(CO)(PPh3)2实现了氢甲酰化反应,且活性和选择性均非常高。该课题组发现,以PPh3为配体的Rh络合物催化氢甲酰化反应可在非常温和的条件下(25 ℃、0.1 MPa H2/CO)实现,反应的选择性(即线性产物与支链产物的摩尔比(l/b))随反应条件的不同而改变。随后Pruett等[8]发现,当过量的PPh3与Rh络合物作用时可得到线性选择性较高的HRh(CO)(PPh3)2催化剂,该催化剂已商品化并在氢甲酰化反应中得到了广泛的应用。
HRh(CO)(PPh3)2催化剂活性物种间的平衡见图2。从图2可看出,HRh(CO)(PPh3)2(2a)在催化体系中很容易发生解离,一分子的PPh3很容易被CO置换从而形成络合物2b;络合物2b的催化活性高,但选择性却不高,其一分子的PPh3继续被一分子的CO置换形成络合物2c,络合物2c的催化活性比络合物2b更高,但选择性最差。因此,在反应时需加入大大过量的PPh3,使平衡向具有高选择性的HRh(CO)(PPh3)2方向移动[8]。但大大过量的配体对后续分离操作不利,同时也不绿色、不经济。
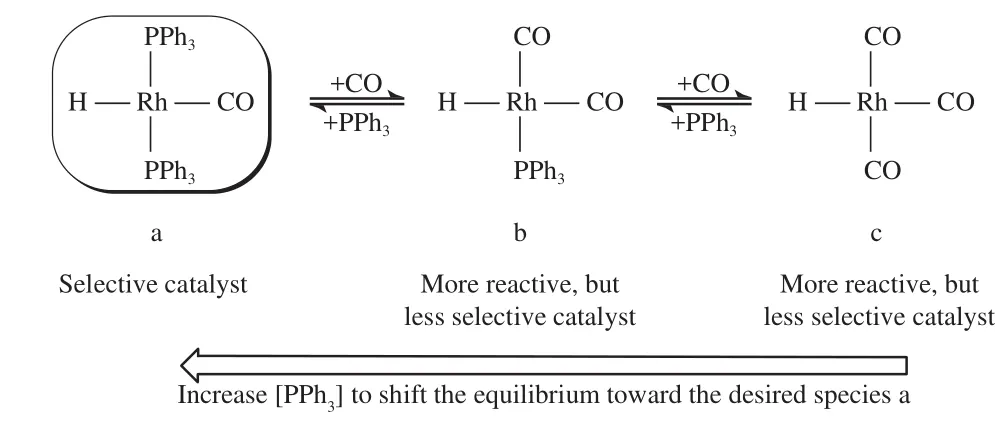
图2 HRh(CO)(PPh3)2催化剂活性物种间的化学平衡Fig.2 Chemical equilibrium between the active species in the HRh(CO)(PPh3)2 catalyst.
2.1 双齿膦配体
为避免单齿膦配体(如PPh3)用量过大的缺点,同时为获得更高的选择性,研究者合成了一系列双齿膦配体。双齿膦配体与Rh的作用远强于相应两个单齿膦配体与Rh的作用,当双齿膦配体与Rh配位时,形成的配位键更加牢固,可避免形成如图2中选择性低的络合物。到目前为止,系列双齿膦配体已被成功合成并应用到线性氢甲酰化反应中,其中,具有代表性的双齿膦配体为Bisbi系列配体[9-13]、Xantphos系列配体[14-16]和具有大位阻的含亚磷酸酯键的Biphephos配体[17-18]。
2.1.1 Bisbi系列配体
Bisbi系列配体最初由Eastman Kodak公司[9]设计合成,是第一例用在Rh催化的氢甲酰化反应中的双齿膦配体,在丙烯的线性氢甲酰化反应中表现出了非常高的选择性(l/b = 30)。此后,一系列Bisbi双齿膦配体被相继合成并成功用于选择性线性氢甲酰化反应,代表性的配体见图3[9-13]。双齿膦配体的骨架及其与金属间螯合的键角对氢甲酰化反应的选择性起决定性作用。Casey等[19-20]对比Bisbi双齿膦配体与其他双齿膦配体时发现,P—Rh—P间的键角决定了线性氢甲酰化反应的选择性。双齿膦配体两种不同的中间体构型见图4。从图4a可看出,当P—Rh—P间的键角接近120°时,有利于两个P原子位于以Rh为中心的正八面体的赤道面上,该构型是形成线性氢甲酰化产物的最佳构型。从图4b可看出,当P—Rh—P间的键角较小时,有利于一个P原子位于以Rh为中心的正八面体的赤道面上,而另一个P原子位于正八面体的一个顶点上,该构型则是形成支链氢甲酰化产物的最佳构型。由于Bisbi双齿膦配体的P—Rh—P间实际的键角为113°,较接近120°,易形成图4a构型,故其氢甲酰化反应生成线性氢甲酰化产物的选择性高。
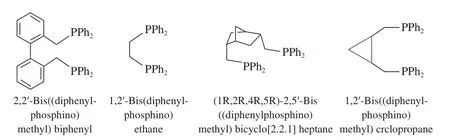
图3 Bisbi系列双齿膦配体Fig.3 Bisibi type bisphosphorus ligands.
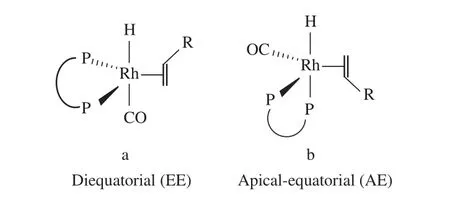
图4 双齿膦配体两种不同的中间体构型Fig.4 Two different reaction intermediates of bidentate ligand.
2.1.2 Xantphos及Naphos系列配体
1990年,Kamer等[21]研究了P—Rh—P间的键角大于99°的配体,并基于此而合成了一系列Xantphos配体(见图5)。
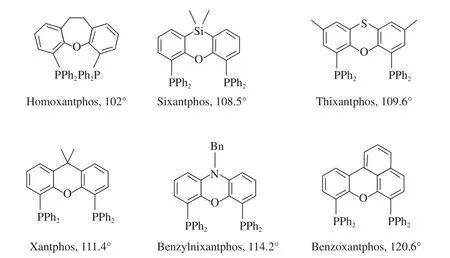
图5 Xantphos系列配体Fig.5 Xantphos type ligands.
该类配体的P—Rh—P间的键角在102°~121°之间,故该类配体在Rh催化的线性氢甲酰化反应中均表现出非常高的选择性,如Benzylnixantphos参与线性氢甲酰化反应时,选择性l/b最高达70。
Klein等[22]报道了含强吸电子取代基的Naphos系列配体(见图6)。该类配体在内烯烃的异构化氢甲酰化反应中表现出很高的选择性(l/b达到9.5)。
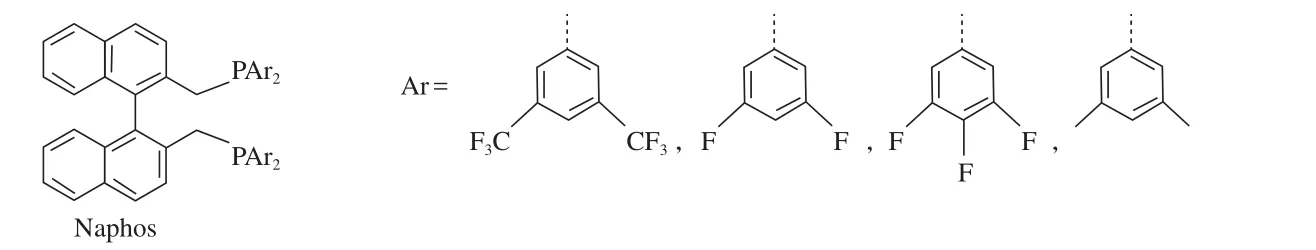
图6 Naphos系列配体Fig.6 Naphos type ligands.
2.1.3 含亚磷酸酯键类型的大位阻配体
含亚磷酸酯键类型的大位阻配体见图7。从图7可看出,该类配体存在结构对称和不对称两种。结构对称的Biphephos配体(见图7a)由Union Carbide公司[17]首次开发,并在氢甲酰化反应中呈现非常高的线性选择性,是优异的线性氢甲酰化配体。采用该类配体时[18],带官能团的烯烃在温和条件下的线性氢甲酰化反应的选择性很高(l/b最高达40),具有非常好的底物普适性,可适用于带羰基、羧基、卤素、乙缩醛基和硫缩醛基等官能团的烯烃。
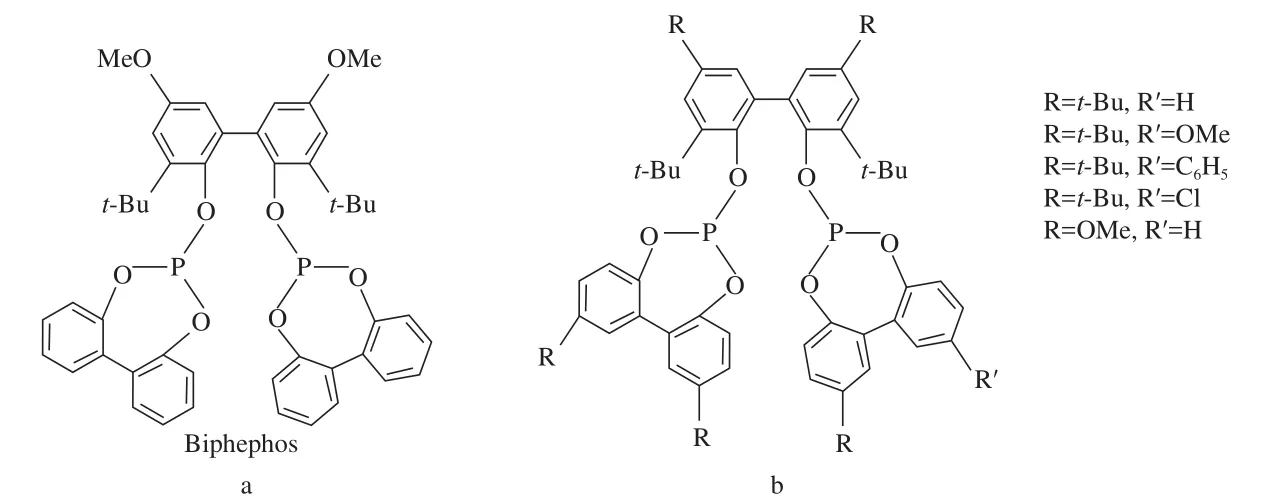
图7 Biphephos系列配体Fig.7 Biphephos type ligands.
van Rooy等[23]设计并合成了结构不对称的Biphephos系列配体(图7b),该类配体为Biphephos配体的衍生化配体,在端烯的线性氢甲酰化反应中也表现出很高的选择性,如在1-壬烯线性氢甲酰化反应中最高得到了l/b=24的选择性。
2.1.4 其他类型的双齿膦配体
在过去数十年里还报道了其他类型的双齿膦配体,其中,具有代表性的双齿膦配体见图8。Magee等[24]开发了含吸电子基团的N-磺胺型的磷酰胺配体(图8a),利用该配体,1-己烯在线性氢甲酰化反应中的选择性l/b=15.8。在Trzeciak等[25]的研究基础上,van der Slot等[26]设计并合成了含吡咯环的双齿膦配体(图8b),利用该配体,端烯在线性氢甲酰化反应中得到了很高的选择性,如1-壬烯在线性氢甲酰化反应中的l/b高达100。近年来,Breit等[27-28]设计并合成了如图8c的配体,该配体由1-羟基-6-二苯磷基吡啶及其羟基被氧化后的异构体通过氢键相互作用得到的。利用该配体,多种类型的端烯在线性氢甲酰化反应中均获得了很高的选择性。Jia等[29]也合成了基于手性螺环骨架的新型双齿膦配体(图8d),相比传统的双齿膦配体,该配体展现出更优异的活性和选择性,当S/C摩尔比为50 000时,该配体在1-辛烯的线性氢甲酰化反应中的选择性l/b高达174.4。
2.2 四齿膦配体
由于双齿膦配体的线性选择性显著高于单齿膦配体,为获得更高的线性选择性,研究者开发了一系列双金属中心四齿膦配体(见图9)。Brousard等[30-32]率先设计并合成了一系列四齿膦配体(图9a~b),该类配体能形成双金属中心Rh络合物。在温和条件下,配体9a和9b均在Rh催化的线性氢甲酰化反应中具有很高的活性和选择性。运用双金属Rh络合物9c,1-己烯在线性氢甲酰化反应中的选择性l/b=33,同时反应最初的转化率达到30 min-1。但将配体9a或9b换为由配体9d或9e形成的双金属中心络合物用于催化氢甲酰化反应,相应的反应速率非常慢,同时获得的选择性也非常低。此外,由双(二苯基膦)甲烷和Rh(nbd)(BF4)2(nbd:降冰片二烯)形成的单金属络合物在线性氢甲酰化反应中也表现出非常慢的反应速率和很低的选择性。表明两个Rh中心在反应中并非单独作用。

图8 其他类型的双齿膦配体Fig.8 Other bisphosphorus ligands.
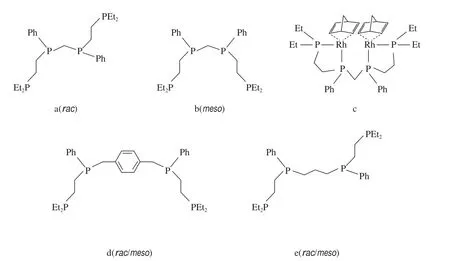
图9 双金属中心四齿膦配体Fig.9 Bimetallic-tetraphosphorus ligands.
本课题组在双齿膦配体的基础上,设计和合成了四齿膦配体(见图10)。配体10a和10b在线性氢甲酰化反应中均表现出了比相应的双齿膦配体更高的选择性[33-34]。采用四齿膦配体10a,1-壬烯在Rh催化的线性氢甲酰化反应中的选择性l/b高达372,1-己烯的选择性l/b达到387。
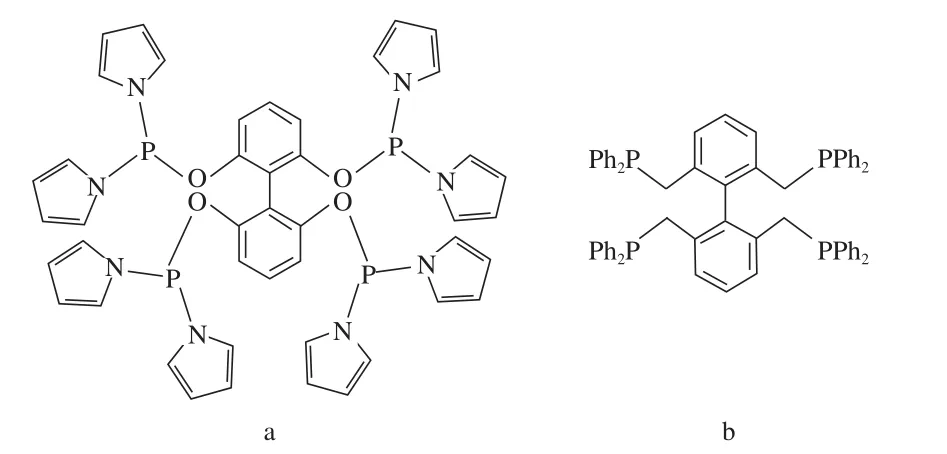
图10 四齿膦配体Fig.10 Tetraphosphorus ligands.
四齿膦配体中P原子与Rh的配位形式有4种(见图11)。从图11可看出,当一个P原子从Rh中心解离下来时,另一个P原子可以重新配位到Rh中心上,因此较双齿膦配体更加有效地抑制了图4中选择性低的催化物种4a或4b的生成,从而增加了Rh中心的膦浓度,在线性氢甲酰化反应中表现出更高的选择性[33]。
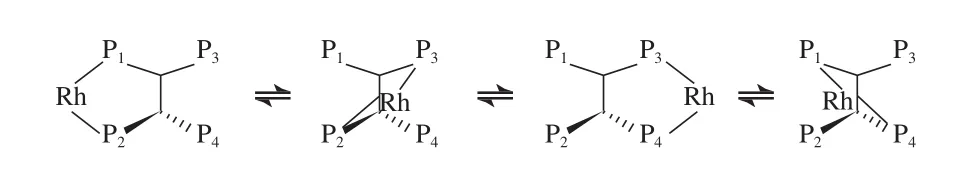
图11 四齿膦配体中P原子与Rh的配位形式Fig.11 Coordination modes of Rh with tetraphosphorus ligands.
2.3 三齿膦配体
Chen等[35-36]开发出一类新型三齿膦配体(见图12)。三齿膦配体与Rh有两种等价的配位模式(见图13),由于Rh中心的膦浓度大幅增加,较传统的二齿膦配体具有更强的螯合能力,因此在线性氢甲酰化反应中应表现更高的选择性。在1-辛烯的线性氢甲酰化反应中,配体12a的线性选择性l/b高达471,而相应的二齿膦配体的线性选择性l/b仅有125;120 ℃下配体12b的线性选择性l/b达66.8,而相应的二齿膦配体的线性选择性l/b仅有47.5。
该课题组还将三齿膦配体应用于混合丁烯的线性氢甲酰化反应中,首次成功地实现了混合烯烃的高选择性氢甲酰化反应,相应的氢甲酰化反应结果见表1。从表1可看出,三齿膦配体在混合丁烯氢甲酰化反应中表现出很高的活性和选择性,其线性产物的含量高达95.5%(x),所得产物正戊醛在工业中经过如图14所示的转化,即可制备新型增塑剂。由于该方法使用的原料为石油化工的废弃物混合丁烯,因而具有成本低的优点。同时,所制备的增塑剂相比传统的增塑剂,由于相对分子质量更高,渗透性较低,因而具有毒性小的优点。
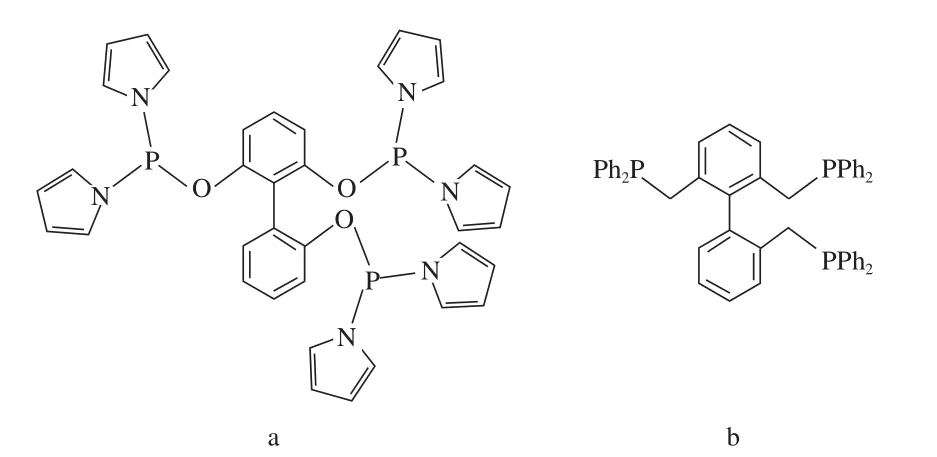
图12 新型三齿膦配体Fig.12 New triphosphorus ligands(Tribi).
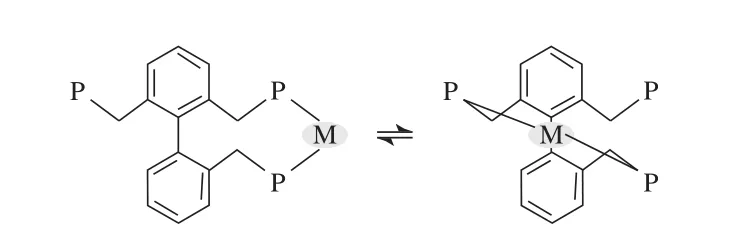
图13 三齿膦配体的两种等价的配位模式Fig.13 Two identical coordination modes of Tribi with rhodium.

表1 三齿膦配体的氢甲酰化反应结果Table 1 Linear hydroformylation with Tribi
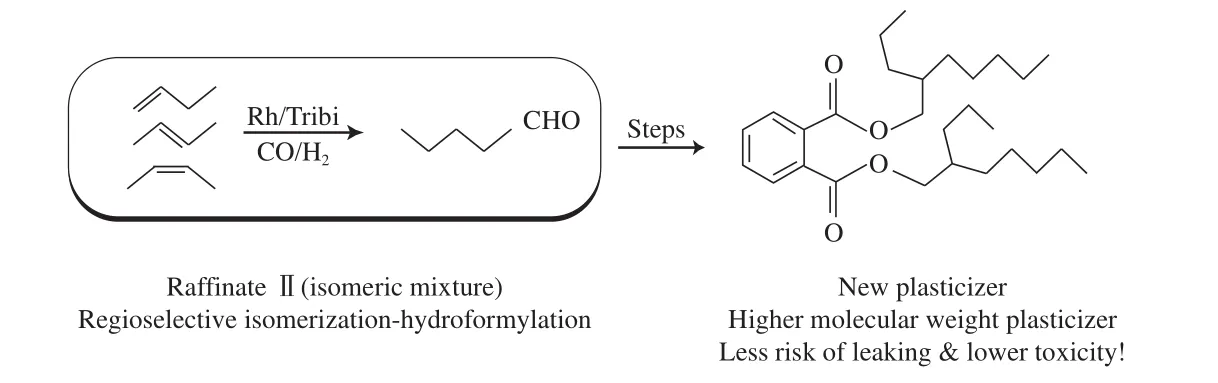
图14 应用三齿膦配体制备新型增塑剂Fig.14 Preparation of a new plasticizer with Tribi.
3 结语
氢甲酰化反应自1938年发现以来,经过几十年的发展,已成为当今世界上规模最大的均相催化反应之一。同时,相应的配体从单齿膦配体到四齿膦配体也有了很大的发展。Bisbi系列、Xantphos系列、大位阻含亚磷酸酯键的Biphephos系列双齿膦配体,四齿膦配体和三齿膦配体是主要用于烯烃选择性线性氢甲酰化反应的配体。近年来发展的三齿膦配体和四齿膦配体较双齿膦配体而言,由于能提供多个螯合位点,能更有效地抑制低选择性的催化活性物种的形成,因而在线性氢甲酰化反应中的选择性较双齿膦配体更高。
尽管线性氢甲酰化的配体已有了很大的进展,但目前发展的膦配体在选择性和底物的适用范围上仍有局限。同时,许多二齿、三齿和四齿膦配体等适用于高效高选择性的氢甲酰化反应的膦配体都已被国外专利保护。而且我国的氢甲酰化工业技术仍依赖国外技术。因此打破国外氢甲酰化技术的垄断,开发具有自主知识产权、高效、高选择性的线性氢甲酰化配体仍然是我国氢甲酰化工业面临的难题,这一问题的解决对提高推动我国线性氢甲酰化工业的发展具有重要的意义。
[1]Pino P,Piacenti F,Bianchi M. Organic Syntheses via Metal Carbonyls[M]. New York:Wiley,1977.
[2]Claver C,van Leeuwen P W N M. Rhodium-Catalyzed Hydroformylation[M]. Dordrecht:Kluwer Academic,2000.
[3]Ungvary F. Application of Transition Metals in Hydroformylation Annual Survey Covering the Year 2004[J]. Coord Chem Rev,2005,249(24):2946 - 2961.
[4]Franke R,Setle D,Böerner A. Applied Hydroformylation[J].Chem Rev,2012,112(11):5675 - 5732.
[5]Roelen O. Organic Carbonyl Compounds Such as Aliphatic Aldehydes:US,2327066[P]. 1943-08-17.
[6]Breit B,Seiche W. Recent Advances on Chemo-,Regio- and Stereoselective Hydroformylation[J]. Synthesis,2001(1):1 - 36.
[7]Evans D,Osborn J A,Wilkinson G. Hydroformylation of Alkenes by Use of Rhodium Complex Catalysts[J]. J Chem Soc,1968(12):3133 - 3142.
[8]Pruett R L,Smith J A. Low- Pressure System for Producing Normal Aldehydes by Hydroformylation of α- Olef i ns[J]. J Org Chem,1969,34(2):327 - 330.
[9]Eastman Kodak Company. Chelate Ligands for Low Pressure Hydroformylation Catalyst and Process Employing Same:US,4694109[P]. 1987-09-15.
[10]Herrmann W A,Kohlpaintner C W,Herdtweck E,et al.Structure and Metal Coordination of the Diphosphane 2,2′- Bis((Diphenylphosphino)Methyl)- 1,1′- Biphenyl (″BISBI″)[J]. Inorg Chem,1991,30(22):4271 - 4275.
[11]Casey C P,Whiteker G T,Melville M G,et al. Diphosphines with Natural Bite Angles Near 120° Increase Selectivity for N- Aldehyde Formation in Rhodium- Catalyzed Hydroformylation[J]. J Am Chem Soc,1992,114(14):5535 - 5543.
[12]Herrmann W A,Schmid R,Kohlpaintner C,et al. Structure and Metal Coordination of the Diphosphine 2, 2′- Bis((Diphenylphosphino)Methyl)- 1, 1′- Binaphthyl (NAPHOS)[J].Organometallics,1995,14(4):1961 - 1968.
[13]Casey C P,Paulsen E L,Beuttenmueller E W,et al. Electron Withdrawing Substituents on Equatorial and Apical Phosphines Have Opposite Effects on the Regioselectivity of Rhodium Catalyzed Hydroformylation[J]. J Am Chem Soc,1997,119(49):11817 - 11825.
[14]Kranenburg M,van der Burgt Y E M,Kamer P C,et al.New Diphosphine Ligands Based on Heterocyclic Aromatics Inducing Very High Regioselectivity in Rhodium- Catalyzed Hydroformylation:Effect of the Bite Angle[J]. Organometallics,1995,14(6):3081 - 3089.
[15]vander Veen L A,Boele M D,Bregman F R,et al. Electronic Effect on Rhodium Diphosphine Catalyzed Hydroformylation:The Bite Angle Effect Reconsidered[J]. J Am Chem Soc,1998,120(45):11616 - 11626.
[16]Carb J J,Maseras F,Bo C,et al. Unraveling the Origin of Regioselectivity in Rhodium Diphosphine Catalyzed Hydroformylation. A DFT QM/MM Study[J]. J Am Chem Soc,2001,123(31):7630 - 7637.
[17]Union Carbide Corporation. Transition Metal Complex Catalyzed Processes: US, 4769498[P]. 1988-09-06.
[18]Cuny G D,Buchwald S. Practical,High- Yield,Regioselective,Rhodium- Catalyzed Hydroformylation of Functionalized α-Actica[J]. J Am Chem Soc,1993,115(5):2066 - 2068.
[19]Casey C P,Whiteker G T,Campana C,et al. Pentacoordinate Iron Tricarbonyl Complexes of Diphosphine Ligands with Bite Angles Greater Than 120°[J]. Inorg Chem,1990,29(18):3376 - 3381.
[20]Casey C P,Whiteker G T,Melville M G,et al. Diphosphines with Natural Bite Angles Near 120° Increase Selectivity for N- Aldehyde Formation in Rhodium- Catalyzed Hydroformylation[J]. J Am Chem Soc,1992,114(14):5535 - 5543.
[21]Kamer P C J,van Leeuwen P W N M,Reek J N H. Wide Bite Angle Diphosphines:Xantphos Ligands in Transition Metal Complexes and Catalysis[J]. Acc Chem Res,2001,34(11):895 - 904.
[22]Klein H,Jackstell R,Wiese K D,et al. Highly Selective Catalyst Systems for the Hydroformylation of Internal Olef i ns to Linear Aldehydes[J]. Angew Chem,Int Ed,2001,40(18):3408 - 3411.
[23]van Rooy A,Kamer P C J,van Leeuwen P W N M,et al.Bulky Diphosphite- Modified Rhodium Catalysts:Hydroformylation and Characterization[J]. Organometallics,1996,15(2):835 - 847.
[24]Magee M P,Luo W,Hersh W H. Electron- Withdrawing Phosphine Compounds in Hydroformylation Reactions:Ⅰ.Syntheses and Reactions Using Mono- and Bis(p- Toluenesulfonylamino)Phosphines[J]. Organometallics,2002,21(2):362 - 372.
[25]Trzeciak A M,Głowiak T,Grzybek R,et al. Novel Rhodium Complexes with N-Pyrrolylphosphines:Attractive Precursors of Hydroformylation Catalysts[J]. J Chem Soc,Dalton Trans,1997(11):1831 - 1838.
[26]van der Slot S C,Duran J,Luten J,et al. Rhodium- Catalyzed Hydroformylation and Deuterioformylation with Pyrrolyl- Based Phosphorus Amidite Ligands:Influence of Electronic Ligand Properties[J]. Organometallics,2002,21(19):3873 - 3883.
[27]Breit B,Seiche W. Hydrogen Bonding as a Construction Element for Bidentate Donor Ligands in Homogeneous Catalysis:Regioselective Hydroformylation of Terminal Alkenes[J]. J Am Chem Soc,2003,125(22):6608 - 6609.
[28]Seiche W,Schuschkowski A,Breit B. Bidentate Ligands by Self- Assembly Through Hydrogen Bonding:A General Room Temperature/Ambient Pressure Regioselective Hydroformy-lation of Terminal Alkenes[J]. Adv Synth Catal,2005,347(11/13):1488 - 1494.
[29]Jia Xiaofei,Wang Zheng,Xia Chungu,et al. Spiroketal-Based Phosphorus Ligands for Highly Regioselective Hydroformylation of Terminal and Internal Olef i ns[J]. Chem Eur J,2012,18(48):15288 - 15295.
[30]Brousard M E,Juma B,Train S G,et al. A Bimetallic Hydroformylation Catalyst:High Regioselectivity and Reactivity Through Homobimetallic Cooperativity[J]. Science,1993,260(5155):1784 - 1788.
[31]Matthews R C,Howell D K,Peng W J,et al. Bimetallic Hydroformylation Catalysis:In Situ Characterization of a Dinuclear Rhodium(Ⅱ)Dihydrido Complex with the Largest Rh- H NMR Coupling Constant[J]. Angew Chem,Int Ed,1996,35(19):2253 - 2256.
[32]Aubry D A,Bridges N N,Ezell K,et al. Polar Phase Hydroformylation:The Dramatic Effect of Water on Mono- and Dirhodium Catalysts[J]. J Am Chem Soc,2003,125(37):11180 - 11181.
[33]Yan Yongjun,Zhang Xiaowei,Zhang Xumu. A Tetraphosphorus Ligand for Highly Regioselective Isomerization- Hydroformylation of Internal Olef i ns[J]. J Am Chem Soc,2006,128(50):16058 - 16061.
[34]Yu Shichao,Zhang Xiaowei,Yan Yongjun,et al. Synthesis and Application of Tetraphosphane Ligands in Rhodium- Catalyzed Hydroformylation of Terminal Olefins:High Regioselectivity at High Temperature[J]. Chem Eur J,2010,16(16):4938 - 4943.
[35]Chen Caiyou,Qiao Yu,Geng Huiling,et al. A Novel Triphosphoramidite Ligand for Highly Regioselective Linear Hydroformylation of Terminal and Internal Olefins[J]. Org Lett,2013,15(5):1048 - 1051.
[36]Chen Caiyou,Li Pan,Hu Zhoumi,et al. Synthesis and Application of a New Triphosphorus Ligand for Regioselective Linear Hydroformylation:A Potential Way for the Stepwise Replacement of PPh3for Industrial Use[J]. Org Chem Front,2014,1(8):947 - 951.
