聚氨酯弹性体中NG和BTTN迁移的介观模拟
2015-05-10虞振飞付小龙蔚红建覃光明谭惠民崔国良
虞振飞, 付小龙, 蔚红建, 覃光明, 谭惠民, 崔国良
(1. 北京理工大学材料学院, 北京 100081; 2. 西安近代化学研究所, 陕西 西安 710065; 3. 航天科技集团科技委, 北京 100048)
1 引 言
在工程应用中,硝酸酯增塑的聚氨酯弹性体存在硝酸酯增塑剂的迁移现象,直接影响着含能材料的使用性能,如硝酸酯增塑聚醚推进剂(NEPE)的能量性能、力学性能、燃烧性能、贮存性能及推进剂与衬层的界面粘接性能[1]。一般情况下,采用浸渍法、气相色谱、高效液相色谱仪和等离子发射光谱等手段分析推进剂和衬层中硝酸酯增塑剂的含量,可以了解硝酸酯增塑剂在推进剂和衬层中的迁移情况[2-4],但实验测定方法需要较长的周期,且具有一定的危险性。
在进行高分子材料的应用研究时,运用分子模拟技术不但可以观察到实验难以测定的现象,还能缩短研究周期,降低研究成本[5]。针对固体推进剂所用粘合剂中增塑剂的迁移和扩散现象,微观尺度上的分子动力学模拟计算获得了较好的结果,如端羟基聚丁二烯中非极性增塑剂的迁移和扩散[6-7]、聚氨酯中硝化甘油的扩散性能[8]等。然而,分子动力学模拟计算方法仅适用于含有数万个原子的体系,可观察的时间范围也仅为皮秒至纳秒,在研究实际问题时难以反映其全貌。最近十几年蓬勃发展起来的耗散粒子动力学DPD(dissipative particle dynamics)属于介观模拟方法,其模拟体系最多可以包含106个原子,在微观尺度的快速动力学和宏观尺度的慢速热力学之间架起了联系的桥梁,能研究微秒范围内聚合物-溶剂体系的相行为[9-10]。但在含能材料领域,有关DPD研究的文献报道较少,如NEPE推进剂粘合剂与增塑剂间相分离[11]、TATB基高聚物粘结炸药(PBX)的介观结构及其演变过程[12]等,而有关固体推进剂或混合炸药所用粘合剂与增塑剂迁移和扩散的DPD研究尚未见到公开的文献报道。
鉴于此,本研究运用分子模拟软件Materials Studio,针对聚氨酯弹性体中丙三醇三硝酸酯(NG)、1,2,4-丁三醇三硝酸酯(BTTN)的迁移问题进行DPD模拟,并将介观模拟计算结果与文献报道的微观模拟计算结果相对照,检验其准确性,并在更大的时间、空间尺度上探索预聚物分子量、温度和硝酸酯含量对NG和BTTN在聚氨酯弹性体中扩散行为的影响,为新型高能固体推进剂和新型混合炸药的研制提供参考。
2 分子模型的建立及分子动力学计算
分子模拟计算面临的首要问题便是如何搭建合适的分子模型。在微观尺度上进行分子动力学计算时,搭建聚合物的三维交联网络已经有了较为成熟的方法[13],而介观尺度下的交联网络模型搭建方法尚未见到公开文献报道。本研究参考DPD相关文献[14],进行一些探索。
本研究所用预聚物端羟基聚氧化乙烯(PEG)、固化剂缩二脲三异氰酸酯(N-100)及聚氨酯的分子结构式如Scheme 1所示,NG和BTTN的分子结构式如Scheme 2所示。
按照文献[14]的方法,将聚氨酯分子的结构式划分为三种片段,如Scheme 3所示。

Scheme1Molecular structure formula of prepolymer, curative, and polyurethane
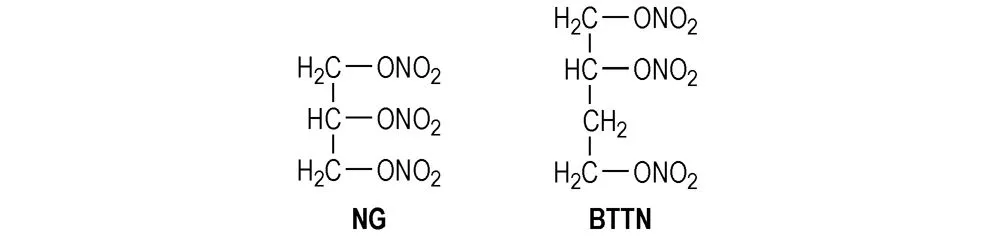
Scheme2Molecular structure formula of NG and BTTN

Scheme3Segments of polyurethane molecule
在Materials Studio的Visualizer模块中分别搭建Scheme 3所示的片段A、B(PEG分子量取5500)、C和两种硝酸酯增塑剂NG、BTTN的分子模型,进行几何优化。具体条件为: COMPASS力场,Materials Studio 内置的Smart minimizer优化方法,范德华作用选择Atom Based,静电作用选择Ewald,位能计算采用球形截断法,cutoff取12.5 Å,spline width取1 Å,buffer width取0.5 Å。
随后,在Amorphous Cell模块中搭建其三维周期边界模型。以片段B为例,其三维周期箱的尺寸为20.28 Å×20.28 Å×20.28 Å,箱内共有878个原子(图1)。在Forcite Plus模块中对优化并弛豫的无定形体系进行分子动力学模拟计算,以恒温恒压系综(NPT)、Anderson恒压器、Berendson恒温器、温度298 K、压力10-4GPa、时间步长1 fs,对优化并弛豫的无定形体系进行200 ps的密度优化,使之接近体系的真实密度。然后,选取恒温恒容系综(NVT),按照上述条件,在真空下进行总模拟时间为200 ps的分子动力学计算,每0.1 ps记录一次全轨迹文件。
图1片段B的三维周期箱
Fig.13D periodic box of segment B
3 结果与讨论
3.1 微观模拟
对图1所示的三维周期箱进行分子动力学计算,所得势能-时间曲线和温度-时间曲线如图2所示。由图2可以看出,在100~200 ps的区间内,其势能和温度波动较小,说明体系达到了平衡。分析相对应的轨迹文件,可以采集各个组分的溶度参数(δ)和密度(ρ)(见表1)。此外,由分子量(Mn)和密度(ρ),可以算出摩尔体积(V)、各组分与NG摩尔体积的比值(R)。这些数据也一并列入表1中。

a. E-t

b. T-t
图2微观体系能量和温度随模拟时间的变化曲线
Fig.2Curves of energy and temperature vs time for micro-scale system
表1各个组分的分子动力学模拟计算结果
Table1Results of molecular dynamics simulation for different components
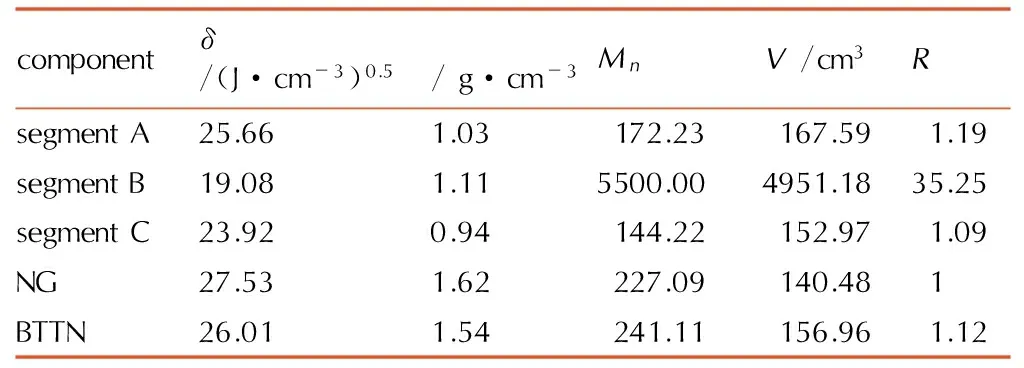
componentδ/(J·cm-3)0.5ρ/g·cm-3MnV/cm3RsegmentA25.661.03172.23167.591.19segmentB19.081.115500.004951.1835.25segmentC23.920.94144.22152.971.09NG27.531.62227.09140.481BTTN26.011.54241.11156.961.12
Note:δis solubility parameter,ρis density,Mnis molecular weight,Vis molar volume,Ris molar volume ratio between component and NG.
3.2 粗粒化的介观模型
为了进行介观模拟(DPD)计算,必须对上述微观分子模型进行粗粒化处理。按照参考文献[14]的方法,将1个NG分子视为1个DPD珠子,根据其余各部分的摩尔体积比值(即表1中的R),可以将 Scheme 3中的片段A、B、C分别视为1、35、1个DPD珠子,1个BTTN分子也视为1个DPD珠子。由此,即可搭建单条聚氨酯分子链的粗粒化模型(见图3a,其中的蓝色珠子为片段A,棕色珠子为片段B,灰色珠子为片段C)。通过粗粒化处理,一个NG、BTTN分子分别由20、23个原子缩减为一个DPD珠子,而一条聚氨酯分子链则从图4中的878个原子缩减为图3a的108个DPD珠子。由表1可知,NG的摩尔体积为140.48 cm3,一个NG分子的体积即为233.36 Å3。按照文献[15]报道的方法,计算出一个DPD珠子的半径为:

a. coarse graining model of polyurethaneb. 3D periodic box of polyurethane/nitrates mixture system
图3介观体系的粗粒化模型
Fig.3Coarse graining models of meso-scale system

(1)
随后,根据实际体系中聚氨酯和硝酸酯的质量分数(NG和BTTN均为6%,聚氨酯为88%),可以搭建聚氨酯/NG/BTTN混合体系的三维周期边界条件模型(见图3b,其中的红色珠子为NG,绿色珠子为BTTN),三维周期箱的尺寸为100×100×100Rc3,即888 Å×888 Å×888 Å,箱内共有15000个DPD珠子。从图3b可以看出,多条聚氨酯分子链与NG、BTTN混合在一起,构成了硝酸酯増塑聚氨酯体系的粗粒化介观模型。
3.3 介观模拟的相关参数
获得表1中各个组分的溶度参数、摩尔体积后,由式(2)可以得到组分之间的Flory-Huggins相互作用参数ij[16],结果见表2。
(2)
式中,δi、δj分别为组分i、j的溶度参数,(J·cm-3)0.5;Vr为参比体积(取组分i、j摩尔体积的平均值,cm3; R为气体常数,8.314 J·(mol·K)-1,T为温度,298 K。
表2组分之间的Flory-Huggins相互作用参数
Table2Flory-Huggins interaction parameters between components

cijABCNGBTTNsegmentA044.700.200.220.01segmentB44.70024.1273.2649.42segmentC0.2024.1200.770.27NG0.2273.260.7700.14BTTN0.0149.420.270.140
由式(3),可得到介观模拟计算所需DPD珠子之间的相互作用参数aij[15],结果见表3。
aij=25+3.50χij
(3)
3.4 DPD计算
介观分子模型搭建好后,根据表3中的aij数据,可在介观模拟计算模块Mesocite中构建相应的DPD力场,并以该力场对图3b所示的聚氨酯/硝酸酯混合体系的介观三维周期箱进行5000步的几何优化,条件为: Smart minimizer优化方法,范德华作用选择Bead Based,静电作用选择Ewald,cutoff取9.24Å,spline width取0,buffer width取0.5 Å。
表3DPD珠子之间的相互作用参数
Table3Interaction parameters of DPD beads

aijABCNGBTTNsegmentA25181.4425.6825.7625.03segmentB181.4425109.42281.40197.96segmentC25.68109.422527.6925.95NG25.76281.4027.692525.49BTTN25.03197.9625.9525.4925
随后进行DPD动力学计算,条件为: 温度298 K、步长500 fs、总模拟时间50000 ps、每1 ps记录一次全轨迹文件。由势能-时间曲线(图4a)和温度-时间曲线(图4b)可以看出,其势能和温度波动较小,说明体系达到了平衡。在40000~50000 ps的区间内,对全轨迹文件进行分析,得到NG、BTTN在聚氨酯/硝酸酯无定形混合物中的MSD-t(均方位移-时间)曲线(图5),经Origin进行线性拟合后得到曲线的斜率。

a. E-t

b. T-t
图4介观体系能量和温度随模拟时间的变化曲线
Fig.4Curves of energy and temperature vs time for meso-scale system

图5介观条件下不同硝酸酯在聚氨酯中的均方位移-时间曲线
Fig.5Mean-square displacement (MSD)-tcurves of different nitrates in polyurethane under the condition of meso-scale
通过爱因斯坦关系式,可求得扩散系数D[13]:
(4)
式中,r(t)代表t时刻粒子的坐标,而r(0)为其初始坐标,尖括号代表对所有原子和初始构型(或时间系综)进行平均,MSD即为均方位移。也就是说,将介观模拟计算获得的MSD-t曲线的斜率除以6,即可得到扩散系数D。
3.5 硝酸酯种类对介观扩散系数的影响
扩散系数是表征分子扩散、迁移能力的重要参数,其含义是沿扩散方向,单位时间内每单位浓度梯度下垂直通过单位面积所扩散某物质的质量或摩尔数[13],其单位为m2·s-1。针对图5中的MSD-t曲线(温度298 K、片段B的分子量5500、NG和BTTN的重量百分含量均为6%),经Origin进行线性拟合后得到曲线的斜率,由式(4)可算出NG和BTTN在聚氨酯中的扩散系数D,见表4。另外,以NG/聚氨酯体系在微观条件下进行分子动力学计算所获得的扩散系数[8]也列入表4中。
表4均方位移-时间曲线的斜率及其对应的扩散系数
Table4Slope ofMSD-tcurves and diffusion coefficient

nitrateslope·1012/m2·s-1D·1012/m2·s-1thisworkref.[8]G10.771.800.65-11.10BTTN1.250.21-
表4中的介观扩散系数与文献所报道相似体系微观尺度下分子动力学模拟计算所获得的扩散系数[8]均处于10-12m2·s-1这一数量级上,证明其结果是可信的。
从表4还可以看出,NG的扩散系数大于BTTN的扩散系数。由图3a来看,聚氨酯分子中片段A、C仅分别为2个、1个DPD珠子,而片段B却为35个DPD珠子,故片段B在聚氨酯分子与硝酸酯的相互作用中起着绝对的主导作用。从图2可以看出,BTTN的分子中比NG多了一个亚甲基,其极性弱于NG,故BTTN的溶度参数值也小于NG而更加接近于片段B的溶度参数值(见表1)。根据高分子物理的溶液理论[16]可知,若溶剂和聚合物的溶度参数分别表示为δ1和δ2,则根据Hildebrand和Scatchard的论点,混合热Hm可由下式得出
ΔHm=V(δ1-δ2)2φ1φ2
(5)
式中,V为溶液的总体积,φ1、φ2分别为溶剂和聚合物的体积分数。聚合物与溶剂混溶性主要取决于混合热,Hm越小则二者越容易混溶。根据(5)式,若δ1和δ2相差越小,Hm就越小,则溶剂与聚合物的混溶性越好。也就是说,可以根据溶度参数差值δ来判断溶剂与聚合物的混溶性。实验结果也证实,若溶度参数相近则聚合物与溶剂易于相溶[17]。由于BTTN与片段B之间的溶度参数差值较小,二者的混溶性比NG-片段B好,故BTTN在聚氨酯中的迁移系数比NG小。而70 ℃加速老化条件下,硝酸酯在NEPE推进剂中的含量测试结果表明,BTTN含量随贮存时间的变化幅度小于NG[1],说明BTTN在NEPE推进剂中的迁移量确实比NG小。
3.6 预聚物分子量对硝酸酯介观扩散系数的影响
预聚物的分子量对聚氨酯弹性体的性能有很大影响。为考察这一参数对NG和BTTN在聚氨酯中扩散系数的影响,在温度298 K、NG和BTTN质量分数均为6%的条件下,选择分子量不同的PEG预聚物(即片段B),按照上述步骤搭建分子模型并进行DPD计算,所得NG和BTTN扩散系数D的变化情况如图6所示。
从图6可以看出,随着预聚物PEG分子量的提高,NG和BTTN在聚氨酯中的扩散系数均增大,且预聚物分子量相同时BTTN的扩散系数依然小于NG的扩散系数。
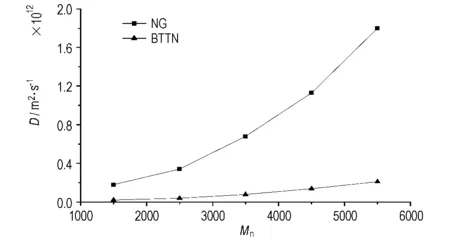
图6预聚物分子量不同时的扩散系数
Fig.6Diffusion coefficient of NG and BTTN with different prepolymers
按照前面提及的模拟方法及步骤进行微观尺度下的分子动力学计算后,可以得到不同分子量预聚物(即片段B)的溶度参数,如图7所示。
由图7可以看出,随着分子量增大,预聚物的溶度参数减小,导致分子量增大后片段B与NG和BTTN的溶度参数差值增大,由式(5)可知片段B与NG和BTTN的混溶性变差,文献报道也证实了这一点[11]。因此,硝酸酯更加容易发生迁移,故二者的扩散系数增大。而相同条件下NG扩散系数大于BTTN的原因,也依然在于其溶度参数不同,前已述及。

图7预聚物分子量不同时的溶度参数
Fig.7Solubility parameters of prepolymers with different molecular weight
3.7 温度对硝酸酯介观扩散系数的影响
为研究温度对硝酸酯在聚氨酯中扩散系数的影响,根据某硝酸酯增塑炸药的实际配方,以片段B的分子量为5500、NG和BTTN的质量分数均为6%的条件,分别在5个温度下进行了DPD计算,所得NG和BTTN扩散系数D的变化情况如图8所示。

图8不同温度下NG和BTTN的扩散系数
Fig.8Diffusion coefficient of NG and BTTN at different temperatures
由图8可以看出,随着温度的升高,NG和BTTN在聚氨酯中的扩散系数也增大,且同一温度下NG的扩散系数仍然大于BTTN。硝酸酯在聚氨酯中的迁移,可理解为硝酸酯与高聚物的相互扩散混合。根据热力学理论,扩散混合的自由能Fm可以表示为
ΔFm=ΔHm-TΔS
(6)
此外,有研究者认为,温度升高后运动单元热运动能量随之升高,聚合物链段间的活动性增加,分子间距离增加,聚合物中的自由体积膨胀、溶胀度升高,使得增塑剂进行扩散的有效空间增大; 且温度升高后,增塑剂分子的活动性增加,加上粘接体系的粘度下降,使得增塑剂的扩散行为更活跃[7]。所以,NG和BTTN在聚氨酯中的扩散系数随温度升高而增大。
3.8 硝酸酯含量对其介观扩散系数的影响
在聚氨酯弹性体中提高硝酸酯的含量,有助于提高固体推进剂或新型混合炸药的能量。为研究NG和BTTN的含量对其在聚氨酯弹性体中扩散系数的影响,以温度298 K、片段B的分子量5500的条件,分别在5种硝酸酯含量下(NG和BTTN的质量分数相同)进行了DPD模拟计算,所得NG和BTTN扩散系数D的变化情况如图9所示。
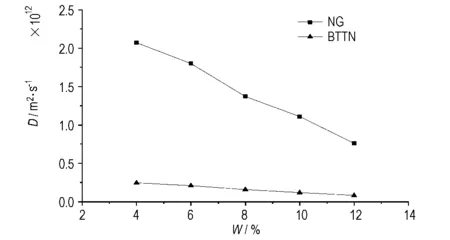
图9硝酸酯含量不同时NG 和BTTN的扩散系数
Fig.9Diffusion coefficient of NG and BTTN with different nitrate contents
从图9可以看出,NG和BTTN在聚氨酯中的扩散系数随其含量的提高而下降,而含量相同时NG的扩散系数大于BTTN。这是因为在交联聚合物的溶胀过程中,存在着2种相反趋势的平衡过程: 增塑剂力图渗入高聚物内部,使高聚物体积膨胀,引起分子网交联点之间分子链的伸展,降低高聚物的构象熵值; 构象熵值降低又必然引起分子网的弹性收缩力,力图使分子网收缩。当弹性体中增塑剂含量超过共容增塑限量后,增塑剂的增加对溶胀度的影响很小,几乎保持不变; 过量的增塑剂占据大量空间,不仅束缚分子网的伸展,而且促使三维分子网络收缩,使其自由体积减少,降低了增塑剂的扩散活动[7]。此外,随NG和BTTN含量的增加,NG和BTTN分子间的相互作用增强,也会降低NG和BTTN在聚氨酯弹性体内孔穴间的跳跃扩散[8]。所以,NG和BTTN的扩散系数出现了下降的趋势。
4 结 论
针对本研究所搭建的粗粒化模型,经过模拟计算后可以得出以下结论。
(1)DPD计算的介观扩散系数与相似体系分子动力学模拟计算的微观扩散系数均处于10-12m2·s-1这一数量级上,证明其结果是可信的。
(2)随着预聚物PEG分子量的提高,NG和BTTN在聚氨酯中的扩散系数增大。
(3)随着温度的升高,NG和BTTN在聚氨酯中的扩散系数也增大。
(4)NG和BTTN在聚氨酯中的扩散系数随其含量的提高而下降。
(5)同样条件下,NG的扩散系数大于BTTN。
(6)介观分子模拟计算结果与文献报道值、理论趋势较为符合,说明图3的硝酸酯/聚氨酯粗粒化模型可以用于考察NG和BTTN在聚氨酯中扩散的情况,这也许是因为迁移、扩散仅仅涉及NG和BTTN分子与聚氨酯分子之间的相溶性所致,且有待于与实验测定数据进一步对比。如欲考察介观条件下该体系的力学性能等,还需进一步探索。
参考文献:
[1] 尹华丽, 李东峰, 王玉. 组分迁移对NEPE推进剂界面粘接性能的影响[J]. 固体火箭技术, 2005, 28(2): 126-129.
YIN Hua-li, LI Dong-feng, WANG Yu. Effect of ingredient migration on interface bonding properties of NEPE propellant[J].JournalofSolidRocketTechnology, 2005, 28(2): 126-129.
[2] 张艳, 王新华, 赵凤起. 硝化甘油向包覆层迁移量的测试方法(浸渍法)研究[J] . 火炸药, 1993(3): 39-43.
ZHANG Yan, WANG Xin-hua, ZHAO Feng-qi. Measurement of migration of nitroglycerine into inhibiting layers/liners[J].ChineseJournalofExplosives&Propellants, 1993, (3): 39-43.
[3] 尹华丽, 王玉, 李东峰. NEPE推进剂粘接体系中的组分迁移及影响[J]. 固体火箭技术, 2009, 32(5): 527-530.
YIN Hua-li, WANG Yu, LI Dong-feng. Ingredient migration and their effect on NEPE propellant bonding system[J].JournalofSolidRocketTechnology, 2009, 32(5): 527-530.
[4] 黄志萍, 谭利敏, 曹庆玮. NEPE推进剂/衬层/绝热层界面迁移组分定量分析[J]. 含能材料, 2010,18(3): 330-334.
HUANG Zhi-ping, TAN Li-min, CAO Qing-wei. Quantitative analysis of migrating components in interface of NEPE propellant/liner/insulation[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2010, 18(3): 330-334.
[5] 杨小震. 分子模拟与高分子材料[M]. 北京: 科学出版社,2002: 258-259.
YANG Xiao-zhen. Molecular Simulation and Macromolecular Materials[M]. Beijing: Science Press, 2002: 258-259.
[6] 李红霞, 强洪夫, 武文明. 丁羟推进剂黏结体系中增塑剂迁移的分子模拟[J]. 火炸药学报,2008,31(5): 74-78.
LI Hong-xia, QIANG Hong-fu, WU Wen-ming. Molecular simulation on plasticizer migration in the bond system of HTPB propellant[J].ChineseJournalofExplosives&Propellants, 2008, 31(5): 74-78.
[7] 李红霞, 强洪夫, 李新其. HTPB推进剂中增塑剂扩散系数计算[J]. 固体火箭技术, 2012, 35(3): 387-390.
LI Hong-xia, QIANG Hong-fu, LI Xin-qi. Measurement of diffusion coefficient of plasticizer in HTPB propellant[J].JournalofSolidRocketTechnology), 2012, 35(3): 387-390.
[8] 王晓, 姚大虎, 白森虎. NG在聚氨酯中扩散性能的分子动力学模拟研究[J]. 含能材料, 2013, 21(5): 594-598.
WANG Xiao, YAO Da-hu, BAI Sen-hu. Molecular dynamics simulation of the diffusion behaviors of NG in polyurethane[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2013, 21(5): 594-598.
[9] Groot, Robert D, Warren, et al. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation[J].TheJournalofChemicalPhysics, 1997, 107(11): 4423-4435.
[10] 陈正隆, 徐为人, 汤立达. 分子模拟的理论与实践[M]. 北京: 化学工业出版社,2007: 240-241.
CHEN Zheng-long, XU Wei-ren, TANG Li-da. Theory and Practice of Molecular Simulation[M]. Beijing: Chemical Industry Press, 2007: 240-241.
[11] LI Song-nian, LIU Yong, TUO Xin-lin. Mesoscale dynamic simulation on phase separation between plasticizer and binder in NEPE propellants[J].Polymer, 2008, 49(11): 2775-2780.
[12] 张艳丽,姬广富,龚自正. TATB基PBX介观结构的耗散粒子动力学模拟[J]. 含能材料, 2008, 16(06): 659-662.
ZHANG Yan-li, JI Guang-fu, GONG Zi-zheng. Dissipative particle dynamics simulation on the mesoscopic structure of TATB-based PBX[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2008, 16(6): 659-662.
[13] 吴超富. 交联环氧树脂的分子模拟研究[D]. 长沙: 湖南大学,2007.
WU Chao-fu. Molecular simulation of crosslinked epoxy resin[D]. Changsha: Hunan University, 2007.
[14] WANG Zhi-gao, LI Jie-hua, TAN Hong. Simulation of self-assembly behaviour of fluorinated phospholipid molecules in aqueous solution by dissipative particle dynamics method[J].MolecularSimulation, 2009, 35(8): 638-647.
[15] Groot, Robert D. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants[J].BiophysicalJournal, 2001, 81(2): 725-736.
[16] 何曼君, 陈维孝, 董西侠. 高分子物理[M].上海: 复旦大学出版社,1990: 114-120.
HE Man-jun, CHEN Wei-xiao, DONG Xi-xia. Polymer Physics[M].Shanghai: Fudan University Press, 1990:114-120.
[17] 李东林, 王吉贵, 仲绪玲. 推进剂中硝化甘油向包覆层迁移的研究[J]. 火炸药学报, 1995(2): 1-4.
LI Dong-lin, Wang Ji-gui, ZHONG Xu-ling. Study of migration of NG in propellant to inhibiting layers/liners[J].ChineseJournalofExplosives&Propellants, 1995(2): 1-4.
