2-亚氨基-1,2-二氢吡啶-1-乙酸的合成
2015-05-10石桂珍由文颖张兆贵孙建梅
石桂珍,由文颖,张兆贵,孙建梅
(潍坊工程职业学院应用化学与生物工程学院,山东青州 262500)
2-亚氨基-1,2-二氢吡啶-1-乙酸则是合成咪唑黄隆的重要中间体,由于除草剂抗性问题的存在,新的除草剂中间体的设计和合成仍是农业化学中一个持续的挑战,日本专利[5]及刘长令[6]报道了该中间体的合成,但对其实验条件未作深入探索。本文在此基础上对实验条件进行了探索,优化了反应条件,以期对其工业化生产提供可利用的数据。
1 实验部分
1.1 试剂及仪器
实验中所用氯乙酸、无水乙醇和三乙胺均为分析纯试剂,2-氨基吡啶为化学纯试剂。
所用仪器包括X-6显微熔点测定仪、Finigan LCQ Advantage型质谱仪、PE-2400型元素分析仪(美国PEKIN-ELMER公司)、Varian INOVA 400 M NMR超导核磁共振波谱仪(美国Varian公司)、AVATAR 360型傅立叶变换红外分光光度计(固体KBr压片)及N-1001型旋转蒸发仪等。
1.2 反应原理
2-氨基吡啶在水和乙醇作溶剂,三乙胺作缚酸剂的条件下,与氯乙酸作用生成2-亚氨基-1,2-二氢吡啶-1-乙酸。
由于氯的电负性较大,而羰基又是吸电子基,所以氯乙酸中的α碳带部分正电荷,该碳原子与吡啶环上具有孤对电子的氮相互吸引形成中间过渡态,最终氯乙酸中的氯夺取氨基上的氢形成氯化氢离去,氨基吡啶发生双键重排得亚胺结构。
1.3 实验过程
将氯乙酸(12.5 g)溶于20 mL水和5 mL无水乙醇的混合溶液中,冷却搅拌下在10~15℃下加入18.5 mL三乙胺,撤去冷却装置,随后加入2-氨基吡啶12.5 g。将反应液加热到75~80℃回流反应5 h后(随着反应的进行,反应液颜色不断加深,由无色到橙红色再到砖红色),向其中加入25 mL无水乙醇(反应液出现分层:上层紫红色,下层橙色沉淀),冰水浴冷却后过滤,并用乙醇(3×20 mL)洗涤滤饼,干燥得白色针状固体16.0 g,收率79.2%(文献值80%[5]),熔点249.5~251.1 ℃(文献值250 ℃[5])。
Anal.(%)C7H8N2O2,found(calcd)C:55.13(55.26),H:5.37(5.26),N:18.33(18.42)。
2 结果与讨论
2.1 FTIR分析
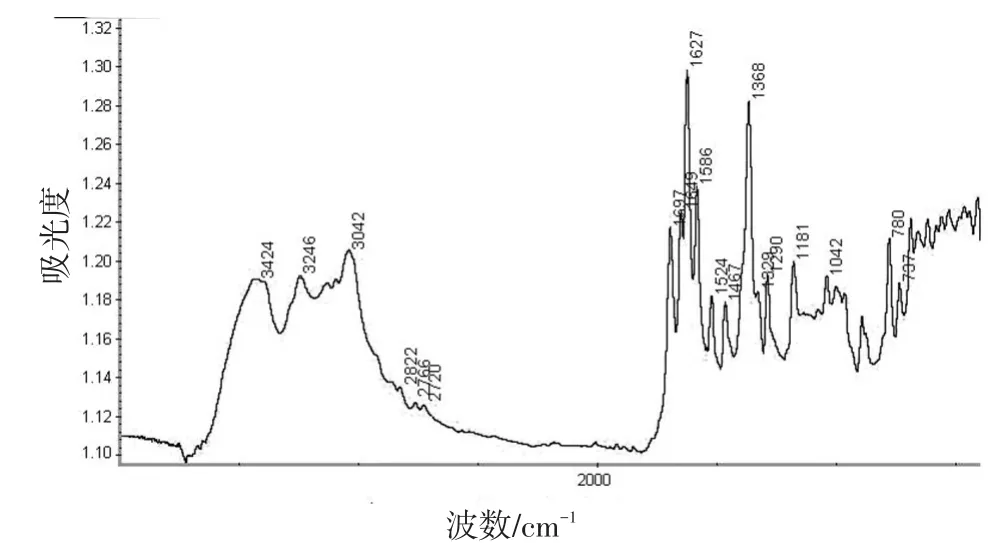
图1 2-亚氨基-1,2-二氢吡啶-1-乙酸的FTIR图
由图1可以看出:3424 cm-1处吸收为胺类N—H伸缩振动;3246 cm-1处吸收为羧酸O—H伸缩振动;3042 cm-1处吸收为芳环C—H伸缩振动;2872 cm-1处吸收为烷烃C—H伸缩振动;1697 cm-1处吸收为羧酸C—O伸缩振动;1649 cm-1处吸收为N—C—C—C共扼伸缩振动;1627 cm-1处吸收为亚胺N—H变形振动;1586、1524 cm-1处吸收为芳环C—C伸缩振动,780 cm-1处吸收为芳环—C—H的变形振动(面外);1368 cm-1处吸收为—CH2—(—CH2—COO—)变形振动;1329 cm-1处吸收为C—N伸缩振动;1293 cm-1处吸收为羧酸C—O伸缩振动与O—H变形振动偶合。与2-氨基吡啶的红外谱图相比[7],其伯胺基3300~3500 cm-1处的两个中强吸收峰转变为3424 cm-1处仲胺的一个中强吸收峰;产物中多了3246 cm-1处羧酸的O—H伸缩振动吸收峰。
2.2 MS分析

图2 2-亚氨基-1,2-二氢吡啶-1-乙酸的MS图
图2为2-亚氨基-1,2-二氢吡啶-1-乙酸的MS图,由于产物的相对分子质量为152,故MS图中的基峰(153.3)为M+1峰。
2.3HNMR分析
以TMS为内标采用Varian INOVA 400 M NMR超导核磁共振波谱仪(美国Varian公司)测得2-亚氨基-1,2-二氢吡啶-1-乙酸的化学位移值为:1H NMR(400 MHz,D2O)4.55(s,2H),6.70(t,J=15.2,1H),6.86(d,J=8.4,1H),7.55(d,J=7.6,1H),7.63(t,J=15.2,1H)。
3 反应条件对产物收率的影响
3.1 反应时间对2-亚氨基-1,2-二氢吡啶-1-乙酸收率的因素
由图3可见,随着反应时间的增加,该反应的收率上升较快,反应2 h收率达到56.3%,4~5 h后收率增加不明显,因此该反应的适宜反应时间为4~5 h。
3.2 反应温度对2-亚氨基-1,2-二氢吡啶-1-乙酸收率的因素
由图4可见,随温度升高,收率呈上升趋势,并且温度越高,收率变化越明显,故该反应的适宜反应温度为该体系的极限温度:75~78℃,此时反应液处于沸腾状态。主要是因为该步反应生成固体,沸腾能够使两相更充分接触,有利于提高产物收率。

图3 反应时间对收率的影响
此外,作者还考察了缚酸剂对产物收率的影响。缚酸剂即捕获酸的试剂,在许多化学农药的合成反应中,如果产物有酸生成又不便及时除去,可加入少量缚酸剂(大部分可回收),来大大提高反应速度和产物转化率[8]。在氨基吡啶与氯乙酸反应的过程中要释放氯化氢,而原料中又有游离的氨基存在,会发生如下副反应:

图4 反应温度对收率的影响

此反应的发生降低了原料的反应活性。因此,在这类反应中一般都要加入一定量的缚酸剂来中和反应生成的氯化氢。常用的缚酸剂有三乙胺、N,N-二甲基苯胺、吡啶等,本实验选择三乙胺为缚酸剂。通过实验比较,不加缚酸剂时副产物明显多于加缚酸剂时的副产物,且产物的颜色较深。不加缚酸剂时收率只有36%左右,加缚酸剂时收率可达70%以上。
3.3 正交试验优选
由前面的探索实验结果,本文选取反应温度(A)为60、70、78 ℃,反应时间(B)分别为2、4、6 h,投料比(C,即氯乙酸与氨基吡啶的物质的量之比)分别为1、1.2、1.5,以乙醇和水作溶剂,加入三乙胺作缚酸剂,正交实验结果如表1所示。

表1 正交实验结果与极差(R)分析
3.3.1 直观分析
极差R的大小反映了因子影响指标的主次关系,由直观分析可以看出,影响该步合成反应收率的主次顺序为:A>B>C;并得到最优实验条件为:反应时间为6 h,投料比为1.5,反应温度为78℃。
3.3.2 方差分析
直观分析不能给出误差的大小,也就不知道结果的精度,而方差检验能够反应数据的波动性,即数据的分散性,方差大小表明数据变化的显著程度,也表明因素对指标影响的大小。氯乙酸与氨基吡啶的物质的量之比对反应收率的影响较小,可并入误差项,计算各均方及F值,列方差分析表见表2。
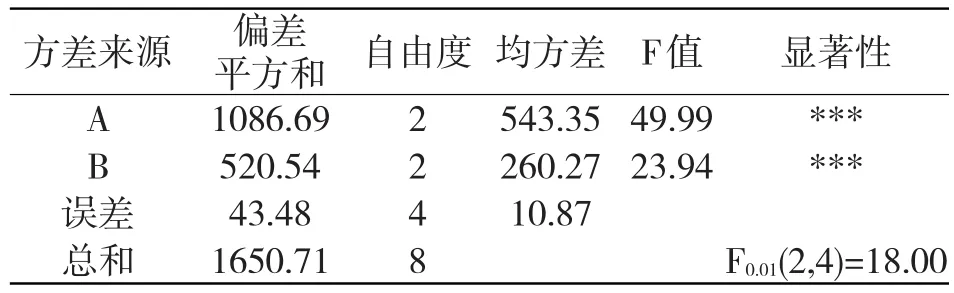
表2 方差分析
经方差分析可知,A、B因素对收率的影响均很显著,且显著性次序为A>B。结合生产实际,从降低成本、节约时间等方面考虑,可取适宜的水平为A3B2C1。
在选取的适宜水平下(反应时间为4 h,反应温度为78℃,氯乙酸与氨基吡啶的物质的量比为1:1)做验证实验,所得产品收率为79.0%,收率较高,且降低了原料成本。
4 结论
4.1 实验制得的2-亚氨基-1,2-二氢吡啶-1-乙酸经分析和表征,与预期结果相符。
4.2 影响2-亚氨基-1,2-二氢吡啶-1-乙酸收率的因素有反应时间、反应温度及缚酸剂等。通过单因素试验、正交试验及方差分析,并结合工业化生产实际,可选取适宜的反应时间为4 h,适宜的反应温度为78℃,氯乙酸与氨基吡啶的物质的量之比为1:1,乙醇和水作溶剂,加入三乙胺作缚酸剂。
4.3 对反应条件的探索为大规模工业化生产提供了可以参考的数据。
[1]TATSUO N,KAZUNARI O,SHIGEYUKI I,et al.Production of condensed heterocyclic rings:JP,1316379[P],1989-12-21.
[2]刘长令.新型稻田除草剂咪唑黄隆[J].农药,1995,34(9):27-30.
[3]阚锦晴,李想,李永舫.聚-2-氨基吡啶电化学合成及性质[J].物理化学学报,2002,18(2):106-111.
[4]刘建超,陈伟志,贺红武.农药化学中的绿色化学[J].化学通报,2004,(10):750-755.
