UPLC-MS/MS法测定3种兽药制剂中违禁药物氯丙嗪
2015-04-27李波平花锦李涵陈谷峰刘能盛肖前李丹赵泉单利君
李波平,花锦,李涵,陈谷峰,刘能盛,肖前,李丹,赵泉,单利君
(1.广东出入境检验检疫局检验检疫技术中心,广州510623;2.山西出入境检验检疫局检验检疫技术中心,太原030024;3.暨南大学包装工程研究所,广东珠海519000)
UPLC-MS/MS法测定3种兽药制剂中违禁药物氯丙嗪
李波平1,花锦2,李涵1,陈谷峰1,刘能盛1,肖前1,李丹1,赵泉1,单利君3
(1.广东出入境检验检疫局检验检疫技术中心,广州510623;2.山西出入境检验检疫局检验检疫技术中心,太原030024;3.暨南大学包装工程研究所,广东珠海519000)
建立了3种兽药制剂中违禁药物氯丙嗪的超高效液相色谱-串联质谱(UPLC-MS/MS)检测方法。用无水乙醇超声提取试样中违禁药物氯丙嗪,采用Waters ACQUITY UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm),乙腈和10 mmol/L乙酸铵+0.1%甲酸溶液流动相,梯度洗脱,电离喷雾电离方式(ESI+),多反应监测(MRM)定量。该方法线性关系良好,相关系数r2达到0.9956,回收率在88.3%~96.7%之间,相对标准偏差(RSD)介于3.7%~8.1%。方法的检出限为0.003 mg/kg。本方法适用于兽药制剂中氯丙嗪违法添加的定性、定量分析。
兽药制剂;氯丙嗪;违禁药物;超高效液相色谱-串联质谱
氯丙嗪是吩噻嗪类代表药物,又名冬眠灵,是常见的安眠、镇静、催眠药物,为中枢多巴胺受体的阻断剂,作用于中枢神经系统,兽医临床上主要被用作镇静药[1]。饲料中添加此类药物可间接起到催肥作用;使用吩噻嗪类药物可降低动物运输过程中的死亡率[2],因此一些不法分子受利益所趋经常使用添加氯丙嗪的兽药。残留的药物通过动物食品进入人体,并经长期蓄积后引起中毒,大剂量时可引起人体肝脏、肾脏的病变,甚至危害生命健康。为此,我国农业部在176号和235号公告中明确禁止在动物饲料中使用氯丙嗪,在动物源食品中不得检出[3-4];农业部第193号公告中,氯丙嗪被明确禁止在兽药中使用。在进出口贸易中,氯丙嗪已被列入禁用类药物,国家规定不得检出,日本肯定列表中也明确表示氯丙嗪不得检出[5]。欧盟早已将氯丙嗪列入禁用清单中[6]。目前,国内外关于氯丙嗪检测的研究涉及饲料[4]、猪肉[1,7]、猪肝[8-9]等动物性食品[3,6]、血液[10]、蛋白[11]、血浆和生物组织[12-14]、尿液[15]等,以生物体内残留检测为主。关于氯丙嗪的检测方法,有气质联用[1,9]、液相色谱[7,11,13]、液质联用[4,6,10]、化学发光[16]等,国内外尚未有测定兽药制剂中氯丙嗪的文献报道。GC和GC-MS对药物的分析需要进行衍生化,耗时较长。LC-MS/MS灵敏度高,选择性和特异性好[4],在检测领域得到越来越广泛的应用。本研究采用乙醇超声提取,建立了UPLC-MS/MS法测定兽药制剂中违禁药物氯丙嗪。
1 材料与方法
1.1 仪器与设备 ACQUITY Ultra Performance LCTM超高效液相色谱仪(美国,Waters公司),Premier串联四极杆质谱仪(美国,Waters公司),HA-180M型电子天平,0.0001 g(日本A&D公司);移液器(德国Eppendorf股份公司):0.2~0.5 mL,1~5 mL,10~100 mL,100~1000 mL;离心机HC-3518(杭州华创科学器材有限公司),涡旋混合器TALBOYS Advanced Vortex Mixer(杭州华创科学器材有限公司),超声波KQ-500DB型(昆山超声仪器有限公司),Mili-Q净化水系统(法国进口),滤膜:0.22 μm(美国Waters公司)。
1.2 药品与试剂
1.2.1 对照品 氯丙嗪标准品,纯度99%,德国Dr.Ehrenstorfer公司。
1.2.2 供试品
1.2.2.1 阴性对照兽药制剂 喹氟宁(粉末)、替米考星(液体)、心肝包治(恩诺沙星可溶性粉),市售,以上三种兽药制剂经检测均不含氯丙嗪。
1.2.2.2 阳性添加兽药制剂 分别按0.01、0.02、0.1 mg/kg的比例在阴性对照兽药制剂中添加氯丙嗪标准物质,混匀。
1.2.2.3 试剂 色谱纯乙腈(美国,Fisher公司),无水乙醇(分析纯),超纯水。
1.2.2.4 标准溶液配制 准确称取氯丙嗪标准品10.0 mg,用乙腈溶解并准确定容到10 mL,配制成浓度为1000 mg/L的标准储备液。该溶液可在0~4℃冰箱中保存12个月。使用时稀释成所需浓度。
1.3 测定方法
1.3.1 质谱分析条件 色谱柱:Waters ACQUITY UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm);流动相:乙腈/10 mmol/L乙酸铵+0.1%甲酸,梯度洗脱;柱温35℃;流速0.3 mL/min;进样量10 μL。梯度洗脱程序见表1。

表1 梯度洗脱程序表
质谱条件如下。电离方式:电喷雾电离(ESI);扫描方式:正离子扫描;离子源温度120℃;脱溶剂气温度380℃;毛细管电压:3.0 kV;锥孔气流:氮气,流速50 L/h;去溶剂气流:氮气,流速700 L/h;碰撞气:氩气,碰撞气压3.5×10-3mbar;检测方式:多反应监测(MRM)。监测条件参见表2。
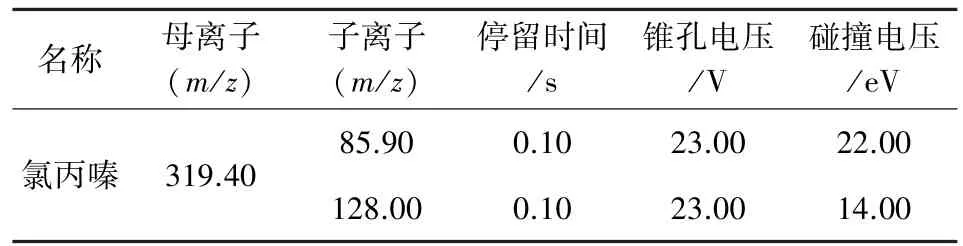
表2 离子对、锥孔电压、碰撞能量表
1.3.2 标准曲线绘制 精密量取标准储备液,用乙腈稀释为0.01、0.02、0.05、0.10、0.20、0.50 mg/L的系列标准溶液,用1.3.1项的质谱条件分析,记录色谱图。
1.3.3 样品前处理 准确称取(2±0.05)g固体或液体样品,置10 mL具塞锥形瓶中,精密移取4 mL无水乙醇摇匀,2800 r/min涡旋3 min,100 W功率下超声提取10 min,静置,经0.22 μm滤膜过滤,收集约1 mL至进样瓶,进行UPLC-MS/MS分析。
1.3.4 样品回收率与精密度 准确称取2.0 g喹氟宁(粉末)、替米考星(液体)、心肝包治(恩诺沙星可溶性粉)于10 mL锥形瓶中,分别添加浓度为0.5 mg/L的氯丙嗪标准溶液0.04、0.08、0.4 mL至样品中,制得添加水平分别为0.01(LOQ)、0.02(2LOQ)、0.1(10LOQ)mg/kg的样品。按照本方法确定的条件进行加标回收重复性试验,每个浓度测定6次,计算回收率及相对标准偏差。
1.3.5 检测限和定量限 取空白样品,按上述步骤操作,测得噪音信号的平均值,按信噪比(S/N≥3)测得方法的检出限(LOD),按信噪比(S/N)≥10测得方法的定量限(LOQ)。
2 结果
2.1 色谱图 采用1.3项的方法测定,待测物质出峰时间为3.11 min,出峰时间短,峰形良好。三种兽药制剂均未检出氯丙嗪。在此仅以喹氟宁为例列图说明。氯丙嗪标准溶液、兽药制剂喹氟宁空白样品、兽药制剂喹氟宁添加氯丙嗪(0.02 mg/kg)样品的色谱图分别见图1、图2、图3。
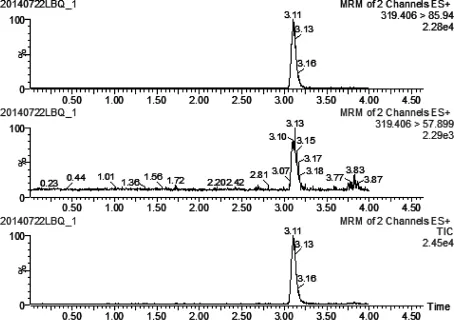
图1 氯丙嗪标准溶液(0.05 mg/L)MRM图
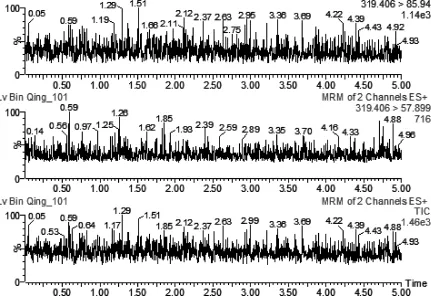
图2 兽药制剂喹氟宁空白样品MRM图
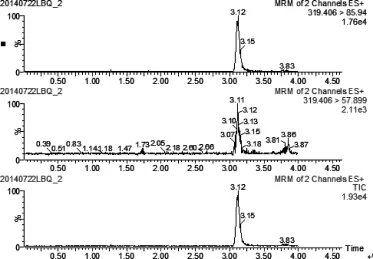
图3 兽药制剂喹氟宁中添加氯丙嗪(0.02 mg/kg)MRM图
2.2 标准曲线 用1.3.1项条件测定1.3.2标准系列溶液,以峰面积为Y轴,对相应的浓度(mg/L)为X轴绘制标准曲线,得到线性方程为:Y=30353X-162.96,相关系数r2=0.9956,线性关系良好。
2.3 方法的回收率与精密度 按照本方法的条件进行加标回收试验,每个浓度水平进行6次重复测定,统计结果见表3。结果表明,三种兽药制剂的添加平均回收率在88.3%~96.7%之间,RSD介于3.7%~8.1%。
2.4 检出限 通过空白样品加标的方法测定,方法的检出限(LOD)为0.003 mg/kg,方法的定量限(LOQ)为0.01 mg/kg。
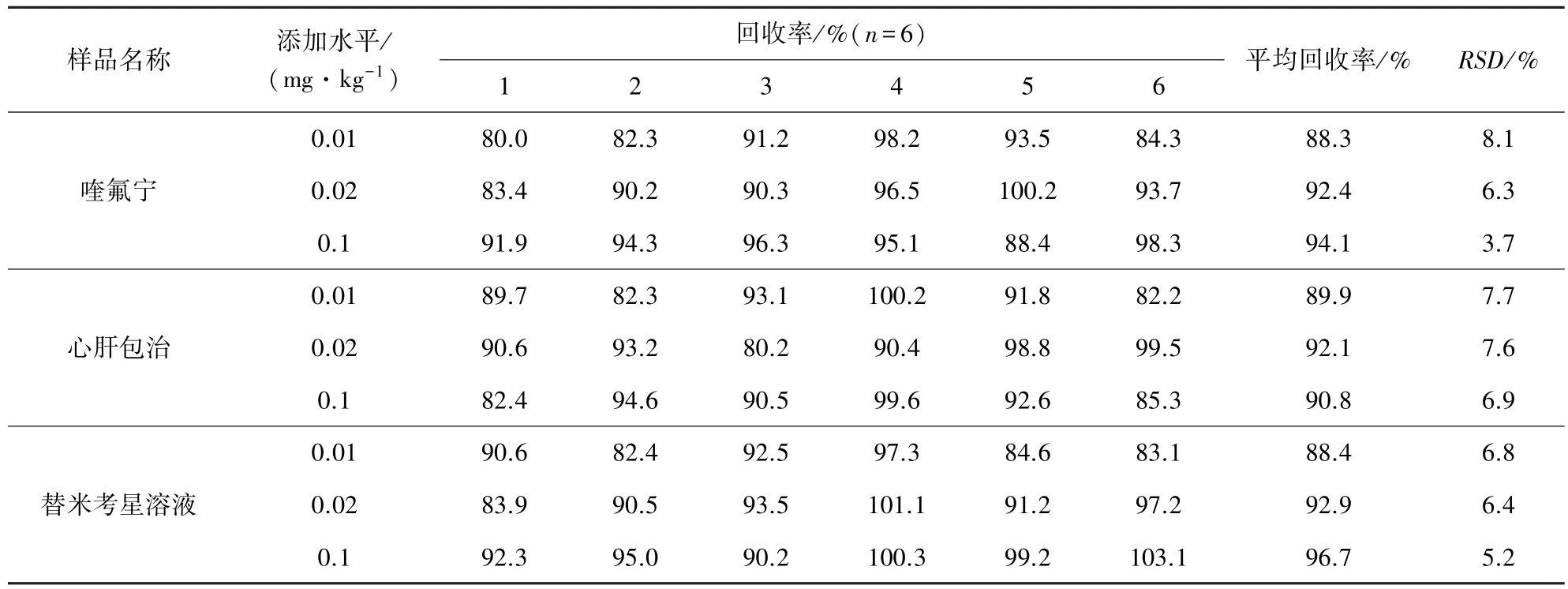
表3 氯丙嗪在兽药制剂中的加标回收率及变异系数表
3 讨论与小结
3.1 提取溶剂的选择 根据兽药典(2000版),氯丙嗪易溶于水、乙醇和氯仿,不溶于乙醚和苯。本文考虑氯仿的毒性较大,且与反相液相色谱流动相的兼容性较差,故排除氯仿。本研究以加标回收率为指标分别比较了水、乙醇、水∶乙醇(1∶1)的提取效果,对添加氯丙嗪浓度为0.01 mg/kg的空白心肝包治进行测定,结果表明,当使用乙醇作为提取剂时目标物回收率最高,因此选用乙醇作为提取溶剂。
3.2 色谱柱及流动相的选择 根据文献[4,10]及实验室现有条件,选用Waters ACQUITY UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm)色谱柱,试验结果表明,采用该柱出峰时间短,与杂峰分离较好。文献[4]提出流动相中加入0.1%甲酸,可降低流动相的pH值,有助于化合物的离子化,可以提高灵敏度。文献[6]提出在流动相中加入一定比例的铵盐,可以使药物易于出峰,改善峰形。乙酸铵是理想的流动相,可有效抑制拖尾,增强电离作用。乙腈和甲醇是液相色谱质谱常用的流动相。综合以上所述,本研究比较了乙腈/10 mmol/L乙酸铵+0.1%甲酸、乙腈/10 mmol/L乙酸铵、乙腈/0.1%甲酸、甲醇/10 mmol/L乙酸铵+0.1%甲酸、甲醇/10 mmol/L乙酸铵、甲醇/0.1%甲酸作为流动相的分离效果。结果表明,选用乙腈/10 mmol/L乙酸铵+0.1%甲酸为流动相,目标化合物的响应较高,离子化效果好,最终选用乙腈/10 mmol/L乙酸铵+0.1%甲酸为流动相。
3.3 质谱条件的优化 根据氯丙嗪物质的结构特征,并参考文献[2-3,10]选择多反应监测方式(MRM)、电喷雾离子源(ESI)进行采集,在正离子扫描模式下进行一级质谱分析,可观察到氯丙嗪的[M+H]+分子离子峰,确定氯丙嗪的准分子离子[M+H]+m/z为319.40。在对准分子离子进行二级质谱扫描时,又得到稳定的碎片离子。进一步对质谱参数锥孔电压和碰撞电压进行优化,最终确定表2中的质谱条件。
本方法为超高效液相色谱串联质谱-电喷雾正离子测定3种兽药制剂中氯丙嗪,该方法操作简单、分析时间短、回收率高,填补了兽药制剂中氯丙嗪检测的空白,为检测兽药制剂中非法添加氯丙嗪提供了有效的技术支持。
[1] 单美娜,徐晓枫,蒲云霞,等.气相色谱质谱法测定猪肉中氯丙嗪残留[J].中国食品卫生杂志,2013,25(5):438-440.
[2] 齐士林,吴敏,严丽娟,等.超高效液相色谱-质谱对动物源食品中氯丙嗪、异丙嗪及其代谢物的测定[J].分析测试学报,2009,28(6):677-681.
[3] 渠岩,路勇,冯楠,等.基质固相分散-超高效液相色谱-串联质谱法同时测定畜禽肉中残留的13中镇静药物[J].食品科学,2012,33(8):252-255.
[4] 许成保,锁然,张峰,等.超高效液相色谱-四级杆飞行时间质谱快速检测动物饲料中10种违禁精神药物[J].色谱,2012,5(30):457-462.
[5] 洪月玲,郝学飞,董柯,等.动物性食品中氯丙嗪残留的液相色谱法检测[J].食品科学,2009,30(14):269-271.
[6] 李丹妮,黄士新,张文刚,等.超高效液相色谱—串联质谱法检测六种动物组织中盐酸氯丙嗪残留[J].中国兽药杂志,2010,44(2):7-11.
[7] 李堃,赵丹莹,郭蒙京.猪肉中氯丙嗪含量的高效液相色谱测定法[J].职业与健康,2013,29(2):181-183.
[8] 李品艾,张会芬,张玲.UPLC法同时测定猪肝中五种磺胺类药物与氯丙嗪残留量[J].医学检验,2010,17(20):77-78.
[9] 吕燕,杨挺,赵健,等.气相色谱-质谱法测定猪肝中氯丙嗪残留量[J].分析试验室,2008,27(3):119-122.
[10]董颖,杜鸿雁,邵永军.UPLC-MS/MS测定全血中的氯丙嗪[J].中国法医学杂志,2013,28(1):44-46.
[11]Jessica J.W.Broeders,Bas J.Blaauboer,Joop L.M.Hermens.Development of a negligible depletion-solid phase microextraction method to determine the free concentration of chlorpromazine in aqueous samples containing albumin[J].Chromatography A,2011,1218:8529-8535.
[12]Guodong Zhang,Alvin V.Terry Jr.Michael G.Bartlett.Sensitive liquid chromatography/tandem mass spectrometry method for the determination of the lipophilic antipsychotic drug chlorpromazine in rat plasma and brain tissue[J].Chromatography B,2007,(854):68-76.
[13]Yadollah Yamini,Mohammad Faraji.Extraction and determination of trace amounts of chlorpromazine in biological fluids using magnetic solid phase extraction followed by HPLC[J].Pharmaceutical Analysis,2014,4(4):279-285.
[14]Hamid Reza Sobhi,Yadollah Yamini,Reza Haji Hosseini Baghdad Abadi.Extraction and determination of trace amounts of chlor⁃promazine in biological fluids using hollow fiber liquid phase microextraction followed by high-performance liquid chromatography[J].Pharmaceutical and Biomedical Analysis,2007,(45):769-774.
[15]Jing CHEN,Chaomei XIONG,Jinlan RUAN.Dispersive Liquidliquid Microextraction Combined with High-performance Liquid ChromatographyfortheDeterminationofClozapineand Chlorpromazine in Urine[J].Huazhong Univ Sci Technol[Med Sci],2011,31(2):277-284.
[16]李旭菲,杨燕英,周考文.毛细管电泳电致化学发光法同时测定氯丙嗪、异丙嗪及其主要代谢物[J].色谱,2012,30(9):938-942.
(编辑:侯向辉)
Determination of Chlorpromazine Illegally Added in 3 Kinds of Veterinary Preparations by UPLC-MS/MS
LI Bo-ping1,HUA Jin2,LI Han1,CHEN Gu-feng1,LIU Neng-sheng1,XIAO Qian1,LI Dan1,ZHAO Quan1,SHAN Li-jun3
(1.Guangdong Exit/Entry Inspection and Quarantine Bureau,Guangzhou 510623,China;2.Shanxi Exit/Entry Inspection and Quarantine Bureau,Taiyuan 030024,China;3.Packaging Engineering Institute of Jinan University,Guangdong,Zhuhai 519000,China)
A method of determination of chlorpromazine illegally added in 3 kinds of veterinary preparations by ultra-performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS)was developed.Chlorpromazine was extracted with ethanol by ultrasonic wave and separated on the Waters ACQUITY UPLC BEH C18 column(50 mm×2.1 mm,1.7μm)using the mobile phase of acetonitrile/mixture of 10 mmol/L ammonium acetate and 0.1%formic acid with gradient elution.Chlorpromazine was detected in electrospray ionization(ESI+)mode using multiple reaction monitoring(MRM).There was a good linear relationship with the correlation rate more than 0.9956.The average recoveries ranged from 88.3%to 96.7%.The standard deviation was between 3.7%and 8.1%.The limit of detection was 0.003 mg/kg.The method was rapid,simple,sensitive and accurate.It was suitable for qualitative and quantitative analysis of the illicit drug chlorpromazine in veterinary preparations.
veterinary preparations;chlorpromazine;illicit drug;ultra-performance liquid chromatographytandem mass spectrometry(UPLC-MS/MS)
2015-02-10
A
1002-1280(2015)05-0032-05
S859.83
质检行业公益性科研专项(201010073-03)
李波平,硕士,工程师,从事进出口商品检测研究。E-mail:lbphappy@163.com
