液质联用法测定猪肝中β-受体激动剂残留的不确定度评定
2015-04-26黄雪英钟锦辉胡麦玲
黄雪英,齐 敏,沈 丹,钟锦辉,胡麦玲,谭 诺
(广州市动物卫生监督所,广州510440)
液质联用法测定猪肝中β-受体激动剂残留的不确定度评定
黄雪英,齐 敏,沈 丹,钟锦辉,胡麦玲,谭 诺
(广州市动物卫生监督所,广州510440)
采用超高效液相色谱-串联质谱法(UPLC-MS/MS)测定猪肝中9种β-受体激动剂残留量,对标准溶液、标准曲线、样品称量、称量重复性、定容体积等因素进行分析,通过评定各不确定度分量及标准不确定度,得出9种β-受体激动剂的合成相对标准不确定度为5.60%~8.90%,影响较大的因素为标准曲线和测量重复性。
不确定度;β-受体激动剂;超高效液相色谱-串联质谱
β-受体激动剂可促进动物生长、抑制脂肪合成,提高瘦肉率,被用于饲养动物后,易在动物组织中残留,从而影响消费者身体健康。农业部明令禁止克仑特罗、沙丁胺醇、莱克多巴胺、氯丙那林等16种β-受体激动剂用于动物饲养。根据农业部1025号公告-18-2008《动物源性食品中β-受体激动剂残留检测液相色谱-串联质谱法》[1],可以同时检测动物源性食品中特布他林、西马特罗、沙丁胺醇、非诺特罗、氯丙那林、莱克多巴胺、克仑特罗、妥布特罗和喷布特罗等9种β-受体激动剂残留量。一个完整的测量结果,除了给出被测量的最佳估计值之外,还应同时给出与该值相关的不确定度。为了更合理、更科学地表示测量结果,依据JJF1059.1-2012《测量不确定度评定与表示》[2]和CNAS-GL06∶2006《化学分析中不确定度的评估指南》[3],本研究建立了一套液相色谱-串联质谱法测定猪肝中β-受体激动剂残留量不确定度的评定方案。
1 材料与方法
1.1 仪器 超高效液相色谱-串联四极杆质谱联用仪(Waters UPLC Xevo TQ);旋涡仪(IKA);固相萃取装置(Supelco);氮吹仪(Organomation);高速离心机(Beckman);振荡器(YAMATO)。
1.2 试剂 乙酸铵、高氯酸、氢氧化钠、乙酸乙酯、叔丁基甲醚、氨水,均为分析纯;甲醇、甲酸,均为色谱纯;实验用水为超纯水。
1.3 标准品 克伦特罗、莱克多巴胺、沙丁胺醇、特布他林、喷布特罗(Dr.Ehrenstorfer GmbH),氯丙那林、妥布特罗(WITEGA)、西马特罗(Sigma Aldrich)、非诺特罗(Toronto Research Chemicals Inc.),纯度均大于98%。
1.3.1 标准储备液(100 μg/mL)的制备 分别准确称取0.01000 g(精确到±0.00001 g)的特布他林、西马特罗、沙丁胺醇、非诺特罗、氯丙那林、莱克多巴胺、克仑特罗、妥布特罗和喷布特罗标准品,用甲醇溶解并定容至100 mL,分别配制成100 μg/mL的标准储备液。
1.3.2 混合标准工作液(0.01 μg/mL)的制备 分别吸取0.1 mL的上述9种β-受体激动剂储备液(浓度100 μg/mL),置于10 mL棕色容量瓶中,用甲醇稀释至刻度,即成浓度为1 μg/mL的混合标准溶液。
吸取0.1 mL浓度为1 μg/mL混合标准储备液,用0.2%甲酸水稀释到10 mL,可得到0.01 μg/mL的混合标准工作液。
1.3.3 空白添加标准曲线的制备 准确称取2 g(精确到0.01 g)空白试样于50 mL离心管内,分别称取 5份。分别向其中添加 50、100、200、400、1000 μL混合标准工作液(浓度0.01 μg/mL),制得浓度为0.25、0.5、1、2、5 μg/kg的空白添加试料。按测试样品处理步骤操作,供超高效液相色谱-串联质谱仪测定。
1.4 测定方法 称取2 g(精确到0.01 g)测试样品,加入乙酸铵缓冲溶液8.0 mL,于37℃避光酶解。酶解后高速离心分出上清液,并用高氯酸调pH后高速离心沉淀蛋白,将上清液转移出来。上清液用NaOH溶液调pH后,用乙酸乙酯10 mL和叔丁基甲醚5 mL分别萃取一次。合并两次萃取有机相,50℃ 下氮气吹干,用2%甲酸溶液溶解,过MCX固相萃取柱(60 mg/3 mL)净化,洗脱液氮气吹干后用0.2%甲酸水溶液1 mL溶解,0.2 μm滤膜过滤后上机测定。
2 测量模型
式中:X—试样中被测物的残留量,单位为微克每千克(μg/kg);
C—样液中被测物的上机测试浓度,单位为纳克每毫升(ng/mL);
V—净化液经氮气吹干后的定容体积,单位为毫升(mL);试验中V=1 mL;
m—供试样品质量,单位为克(g);试验中m=2.00 g。
考虑到实验过程中各种随机因素对测量结果的影响,在测量模型中引入重复性系数frep,建立样品中β-受体激动剂药物残留量不确定度测量模型如下
由测量模型可看出,影响测定结果不确定度urel(X)的主要因素有:由标准曲线计算样液中被测物的浓度C引入的不确定度urel(C),净化液经氮气吹干后定容体积引入的不确定度urel(V),称量样品引入的不确定度urel(m),整个实验重复性(包括提取净化过程和仪器进样)的不确定度通过urel(frep)来体现。
由此得到试样中被测物的残留量的相对标准不确定度urel(X)可由下式计算:

3 相对标准不确定度分量的评定
3.1 实验合成重复性的相对标准不确定度urel(frep)按实验方法平行测定某样品5次,测得样品含量为X1~X5,分别计算各药物平均值和标准偏差测定结果和计算数据见表1。
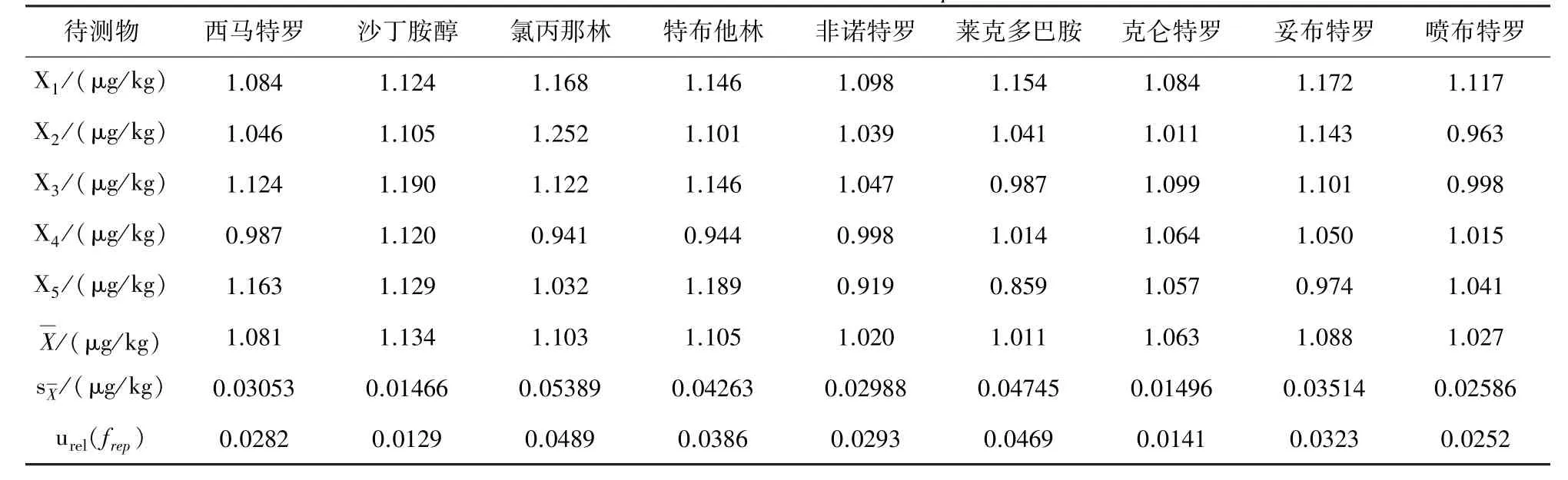
表1 重复性相对标准不确定度urel(frep)计算结果表
3.2 样液中被测物的上机测试浓度C的相对标准不确定度urel(C) 样液中被测物的上机测试浓度C的相对标准不确定度urel(C)由两部分构成,一是制备空白添加标准曲线产生的不确定度urel(Cs),一是标准曲线拟合出样液中被测物的浓度产生的不确定度urel(C0)。
3.2.1 制备空白添加标准曲线产生的不确定度urel(Cs)Cs的不确定度主要来源包括:制备标准储备液引入的不确定度u1(Cs);配制混标工作液引入的不确定度u2(Cs);向空白基质样品中添加标准溶液引入的不确定度u3(Cs)。

3.2.1.1 制备标准储备液引入的相对标准不确定度u1(Cs) 标准储备液的不确定度主要由标准品称量、定容和标准品本身的不确定度决定。
(1)标准品称量引入的相对标准不确定度u11(Cs)
称量标准品的天平最大允许误差为±0.05 mg,取均匀分布,天平称量引入的相对标准不确定度为:

(2)标准液定容引入的相对标准不确定度u12(Cs)
标准品配制使用的100 mL A级容量瓶允差±0.10 mL,取均匀分布,相对标准不确定度:
定容至容量瓶刻度的变动性:重复定容10次,标准偏差为 0.030 mL,相对标准不确定度ur,2(V100mL)=0.030 mL/100 mL=0.0003;
假定实验室温度在20℃±5℃,20℃时甲醇的体积膨胀系数[4]为1.1×10-3/℃,均匀分布,温度引起的相对标准不确定度

相对标准不确定度:
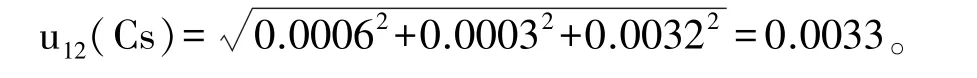
(3)标准品纯度引入的相对标准不确定度u13(Cs)
由供应商(或证书)提供的标准品纯度引入的相对标准不确定度分别为:西马特罗0.005,沙丁胺醇0.005,氯丙那林0.005,特布他林0.01,非诺特罗0.005,莱克多巴胺0.01,克仑特罗0.005,妥布特罗0.005,喷布特罗0.01。
3.2.1.2 制备混标工作液(0.01 μg/mL)引入的相对标准不确定度u2(Cs) 按1.3.2项制备混标工作液(0.01 μg/mL),用0.1 mL移液器吸取甲醇溶液2次,10 mL容量瓶用甲醇和水各定容1次。
(1)10 mL容量瓶导致的相对标准不确定度
10 mL容量瓶的相对标准不确定度计算方式类似3.2.1.1项中(2),允差为±0.02 mL,不确定度为定容重复性标准偏差0.015 mL;温度变动性:溶剂为甲醇时,引起的不确定度为溶剂为水时,引起的不确定度为(20℃时水的体积膨胀系数为2.1×10-4/℃)。
用甲醇配制成浓度为1 μg/mL的混合标准溶液时,10 mL容量瓶引入的相对标准不确定度为
用0.2%甲酸水配制成浓度为0.01 μg/mL的混合标准溶液时,10 mL容量瓶引入的相对标准不确定度为
(2)移液器导致的相对标准不确定度
用移液器导致的体积相对标准不确定度主要有3个分量:校准分量ur,1(V)、充液重复性变化分量ur,2(V)和温度变化分量 ur,3(V),计算方式类似3.2.1.1项中(2)。移液器的容量允许误差和测量重复性参考JJG 646-2006移液器检定规程[5]的要求。
使用移液器引起的体积相对标准不确定度ur(V)计算结果见表2。

表2 使用移液器引起的体积相对标准不确定度表
制备混标工作液(0.01 μg/mL)时,用100 μL移液器移取0.1 mL甲醇溶液两次,引入的相对标准不确定度为:

3.2.1.3 向空白基质样品中添加标准溶液引入的相对标准不确定度u3(Cs) 以添加浓度为1 μg/kg的空白添加标准试样为例,需移取200 μL混合标准工作液(0.01 μg/mL),加入2 g空白试样中制备而得。该体积导致的相对标准不确定度为0.0046(具体数据见表 2)。同理添加成 0.25、0.5、2、5 μg/kg等浓度时相对标准不确定度分别为0.0091、0.0040、0.0041、0.0037。
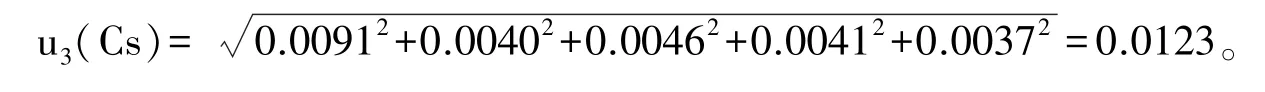
制备标准曲线引入的相对标准不确定度urel(Cs)分别为:西马特罗0.0163,沙丁胺醇0.0163,氯丙那林 0.0163,特布他林 0.0184,非诺特罗0.0163,莱克多巴胺0.0184,克仑特罗0.0163,妥布特罗0.0163,喷布特罗0.0184。
3.2.2 标准曲线拟合过程的相对标准不确定度urel(C0)添加5个不同浓度的混合标准样品0.25、0.5、1、 2、5 μg/kg,按样品处理步骤操作后上机,对应上机浓度0.5、1、2、4、10 ng/mL,每一浓度测定3次。各种化合物以上机浓度C为横坐标,谱图强度峰面积Y为纵坐标,拟合标准曲线,建立数学模型:Yi=bCi+a,计算各化合物线性方程的斜率、截距和相关系数,相关数据见表3。

表3 各化合物回归直线测量结果表
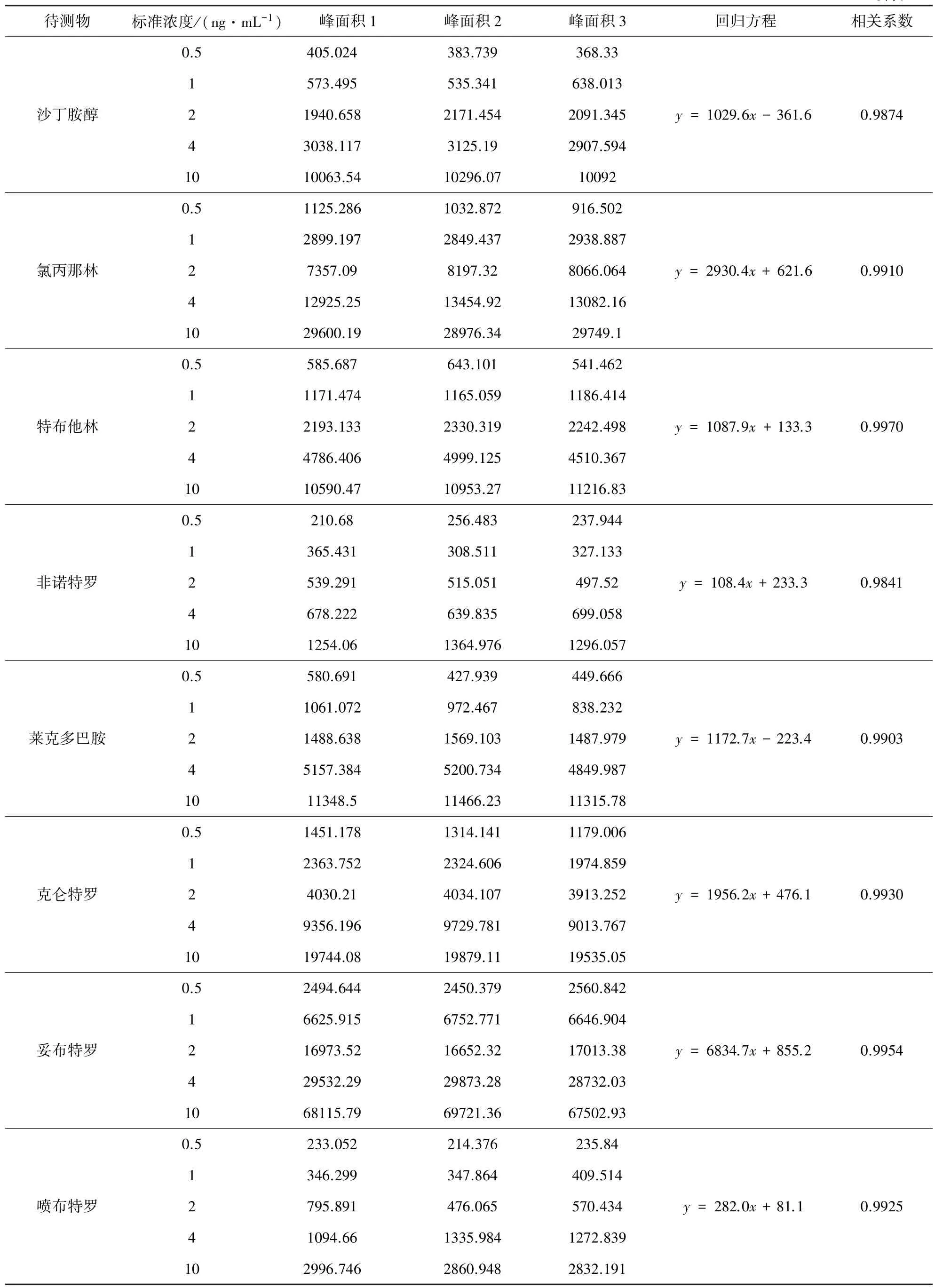
待测物 标准浓度/(ng·mL-1) 峰面积1 峰面积2 峰面积3 回归方程 相关系数沙丁胺醇0.5 405.024 383.739 368.33 1 573.495 535.341 638.013 2 1940.658 2171.454 2091.345 4 3038.117 3125.19 2907.594 10 10063.54 10296.07 10092y=1029.6x-361.6 0.9874氯丙那林0.5 1125.286 1032.872 916.502 1 2899.197 2849.437 2938.887 2 7357.09 8197.32 8066.064 4 12925.25 13454.92 13082.16 10 29600.19 28976.34 29749.1y=2930.4x+621.6 0.9910特布他林0.5 585.687 643.101 541.462 1 1171.474 1165.059 1186.414 2 2193.133 2330.319 2242.498 4 4786.406 4999.125 4510.367 10 10590.47 10953.27 11216.83y=1087.9x+133.3 0.9970非诺特罗0.5 210.68 256.483 237.944 1 365.431 308.511 327.133 2 539.291 515.051 497.52 4 678.222 639.835 699.058 10 1254.06 1364.976 1296.057y=108.4x+233.3 0.9841莱克多巴胺0.5 580.691 427.939 449.666 1 1061.072 972.467 838.232 2 1488.638 1569.103 1487.979 4 5157.384 5200.734 4849.987 10 11348.5 11466.23 11315.78y=1172.7x-223.4 0.9903克仑特罗0.5 1451.178 1314.141 1179.006 1 2363.752 2324.606 1974.859 2 4030.21 4034.107 3913.252 4 9356.196 9729.781 9013.767 10 19744.08 19879.11 19535.05y=1956.2x+476.1 0.9930妥布特罗0.5 2494.644 2450.379 2560.842 1 6625.915 6752.771 6646.904 2 16973.52 16652.32 17013.38 4 29532.29 29873.28 28732.03 10 68115.79 69721.36 67502.93y=6834.7x+855.2 0.9954喷布特罗0.5 233.052 214.376 235.84 1 346.299 347.864 409.514 2 795.891 476.065 570.434 4 1094.66 1335.984 1272.839 10 2996.746 2860.948 2832.191y=282.0x+81.1 0.9925
被测样品2次平行测定后,通过拟合直线求出SR和各化合物浓度C0,根据公式(1)计算出被测样品中所含各待测物残留量X0,按(3)式计算各待测物浓度的标准不确定度u(C0)。

其中Yi—标准溶液的峰面积测定值;
Ci—标准溶液的浓度值;
b—拟合直线的斜率;
a—拟合直线的截距;
n—标准溶液测试次数,本实验中n=15;
p—C0测试次数,本实验中p=2;
C0—由线性方程计算得各化合物浓度;


表4 浓度C的不确定度计算的有关量值表
3.3 定容体积的相对标准不确定度urel(V) 用移液器定容至1 mL,根据表2,urel(V)= 0.0037。
3.4 称量样品的相对标准不确定度urel(m) 称量样品的天平最大允许误差:±5 mg,采用均匀分布,2.00 g=0.0014。
3.5 合成标准不确定度 urel(X)及扩展不确定度Urel(X) 根据公式(2)计算合成标准不确定度,取包含因子k=2,则相对扩展不确定度Urel(X)=k× urel(X),结果见表5。

表5 合成标准不确定度的有关量值表
4 不确定度报告
依据农业部1025号公告-18-2008对猪肝样品中β-受体激动剂残留量进行检测,对样品平行测定2次,取包含因子k=2,测定结果见表6。
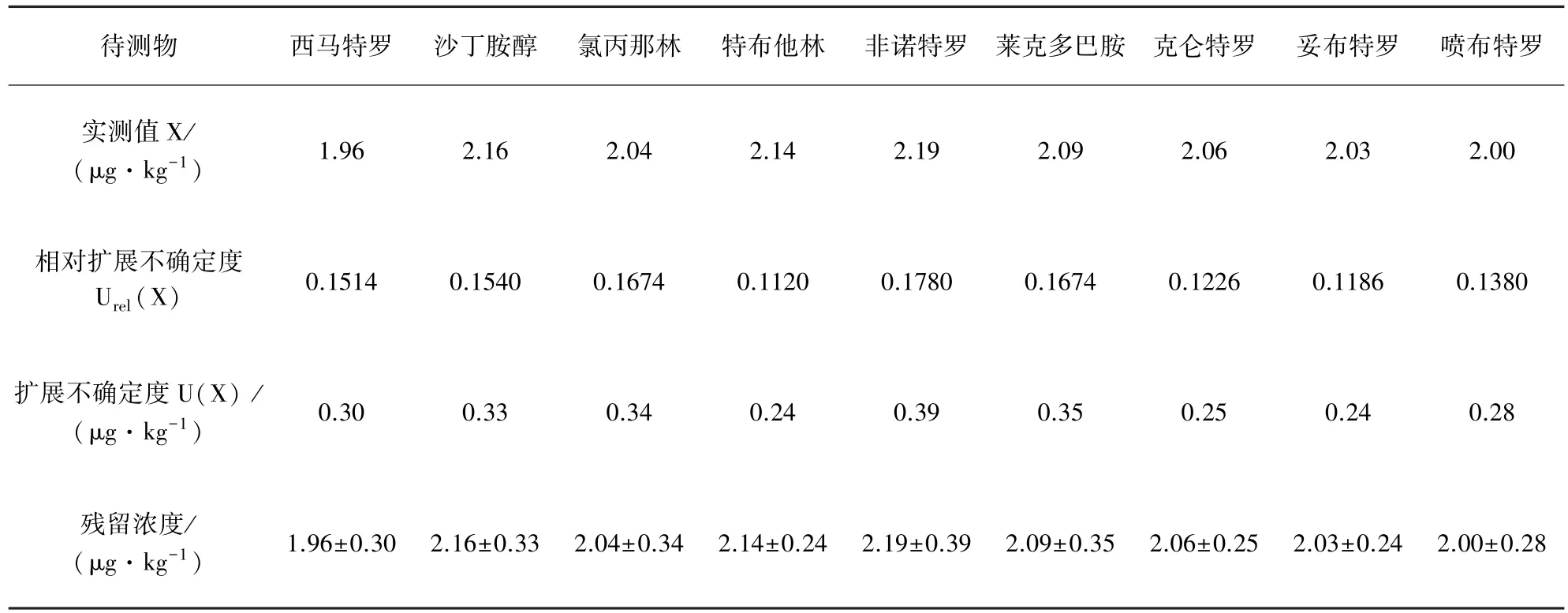
表6 猪肝中β-受体激动剂残留量检测结果表
5 结论
从以上分析数据可以看出,实验过程中,样品称量和最终定容体积引入的不确定度极小,可忽略不计,影响测定结果不确定度的主要因素为标准曲线和样品的测量重复性。所以测试过程中,要严格控制实验条件,保证标准曲线具有良好的线性以及实验具有良好的重复性,才能有效的保证检测结果的准确性。
[1]农业部1025号公告-18-2008.动物源性食品中β-受体激动剂残留检测液相色谱-串联质谱法[S].
[2]JJF1059.1-2012.测量不确定度评定与表示[S].
[3]CNAS-GL06:2006.化学分析中不确定度的评估指南[S].
[4]李兰英,丁 敏,徐 勤,等.LC-MS/MS测定猪尿中盐酸克伦特罗不确定度的评定[J].中国计量学院学报,2012,23(1):7-12.
[5]JJG 646-2006.移液器检定规程[S].
(编 辑:侯向辉)
Uncertainty Evaluation of β-Agonists in Pork Liver by Ultra Performance Liquid Chromatography-tandem Mass Spectrometry
HUANG Xue-ying,QI Min,SHEN Dan,ZHONG Jin-hui,HU Mai-ling,TAN Nuo
(Guangzhou Animal Health Inspection Institute,Guangzhou510440,China)
Evaluation on uncertainty of measurement in the determination of 9 kinds of β-agonists in pork liver by ultra performance liquid chromatography-tandem mass spectrometry was practiced.The main factors,including standard solution,standard curve,weighing of sample,measurement repeatability,and volume were evaluated.The combined uncertainty of 9 kinds of β-agonists was in the range of 5.60%~8.90%.The results showed that the measurement uncertainty was mainly due to the standard curve and measurement repeatability.
uncertainty;β-agonists;ultra performance liquid chromatography-tandem mass spectrometry(UPLCMS/MS)
2014-11-03
A
1002-1280(2015)01-0045-07
S859.84
黄雪英,硕士,从事兽药残留检测技术研究。E-mail:ssherry109@163.com
