常山碱及其类似物关键中间体的合成研究进展*
2015-04-23宋卫强刘丹妮
宋卫强,刘丹妮,陆 群
(西南交通大学生命科学与工程学院,四川成都 610031)
天然产物的特征骨架结构所产生的生物学特性、药理活性和潜在成药性给药物化学家的研究带来灵感,多数天然产物均可通过其特征骨架结构碎片被识别,例如苯二氮卓酮和喹唑啉等结构[1]。3-羟基哌啶(1)作为一种特征片段,广泛存在于多种天然产物中,例如常山碱(2)及其类似物常山酮(3),1-脱氧野尻霉素(4),3-羟基哌啶酸(5),(-)-苦马豆碱(6),(-)-牧豆树宁(7)及(- )-Deoxocassine(8)(Chart 1)等[4-8]。
2和3主要存在于植物常山和伞形秀球等植被中,上世纪50年代Jiang等[9]首次从该植被中分离出(+)-2和(+)-异常山碱,基于氧化和降解实验,Koepfli等[9]提出了常山碱的平面结构。1952年Baker研究小组[4]首次完成了2的化学全合成。由于忽略了2和3之间的差向异构化作用,错误地提出了2的绝对构型。直到二十世纪末,Koboyashi研究小组[4]在不对称全合成2的工作中纠正了Baker小组的错误,提出了2的正确绝对构型和相对构型。这也促使科研工作者们再度产生了对2及其衍生物的合成和临床研究的兴趣。近年来研究发现2及其多种类似物具有非常好的抗疟、抗肿瘤、抗纤维化、抗球虫及抗隐孢子虫等药理活性[9-11]。对2及其类似物的化学合成探索已有50多年的历史,由于片段中间体1合成难度大,成本高,国外只有几家公司在生产,而国内尚未实现生产工业化,主要依托进口和从常山块根提取获得[12]。
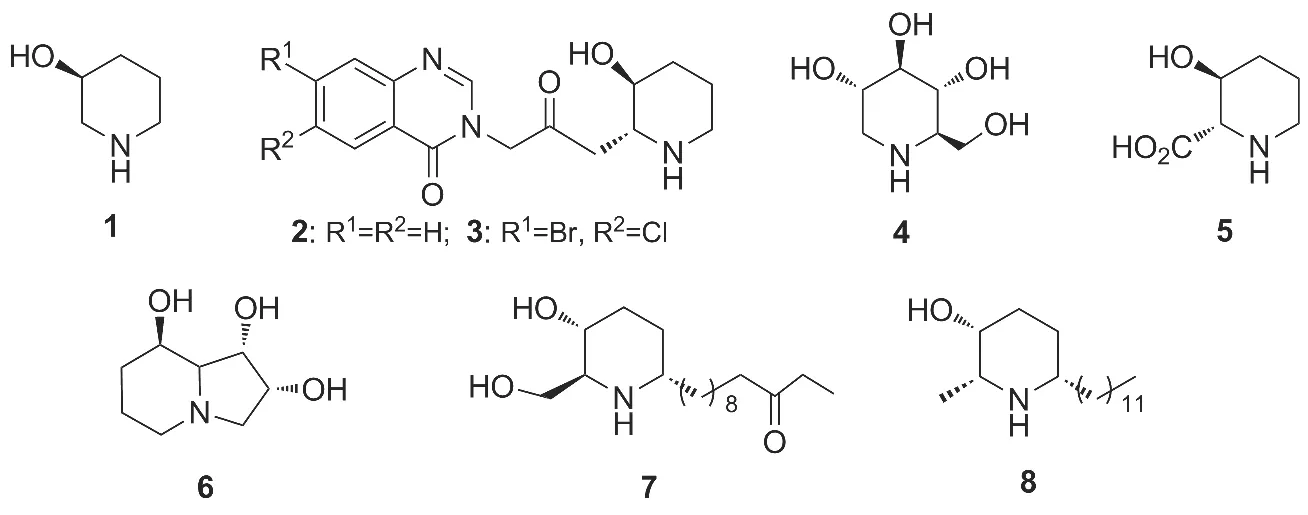
Chart 1
本文从哌啶环构建策略包括吡啶环还原法、分子内亲核进攻法、RCM反应(关环复分解反应)及扩环反应法等出发,综述了近年来2及其类似物关键中间体1的合成研究进展,并对各合成路线的优缺点进行了简要评述。
1 吡啶环还原法
吡啶环还原是构建哌啶环的重要方法。2011年江涛研究小组[13]以 2-甲基-3-羟基吡啶(9)为原料,经甲基化、去质子化、乙腈加成和水解反应制得酮(11);11经Rh/Al2O3催化氢化还原吡啶环制得哌啶酮化合物(12);12经羰基α-H溴代和仲氮保护得哌啶环中间体(13),收率33.9%。13与喹唑啉酮母核缩合,水解得3(Scheme 1),收率11.6%。
该线路是对Baker小组[13]合成线路的优化,比较而言具有操作简单和收率高等优点,但碘甲烷、正丁基锂及Rh/Al2O3等试剂价格昂贵,且碘甲烷毒性较大,工业生产成本依然较高。
2013年吴世林研究小组[14]以3-羟基吡啶(14)为原料,与甲醛和二甲胺经Mannich反应,甲基化及Pd/C催化氢化还原制得16;16经氧化和Cope消除的串联反应,臭氧化生成化合物17,接着与丙酮缩合后再经氢化合成2-丙酮基-3-甲氧基哌啶(19,Scheme 2),收率35.8%。
该线路所用原料均为大宗商品,廉价易得;所涉及的反应均为经典反应,收率较高、反应条件温和、操作简单,易于实现工业化。

Scheme 1
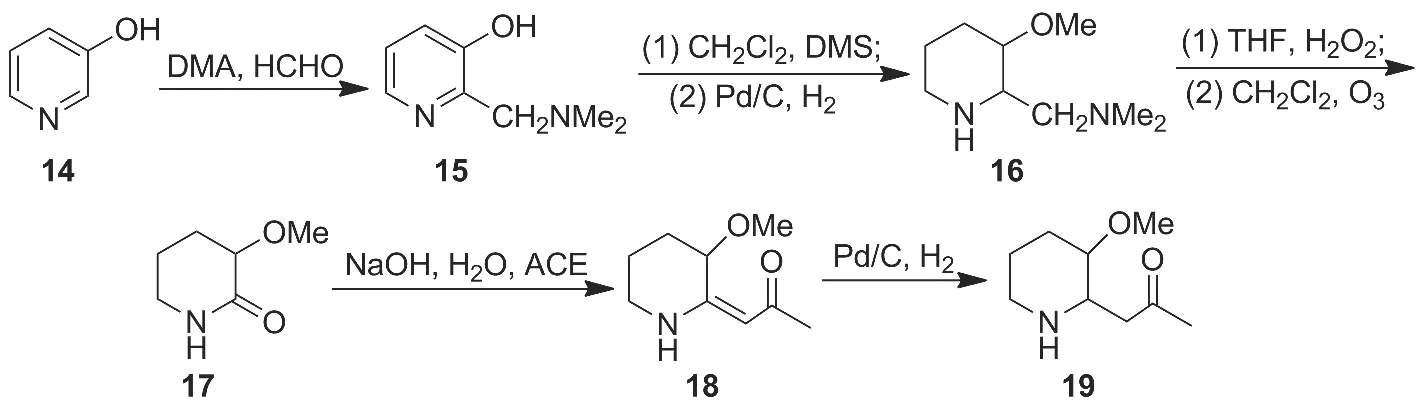
Scheme 2
2 分子内亲核进攻法
2.1 还原胺化法
分子内醛基或酮羰基与胺发生还原胺化反应是构建氮杂环的一个重要手段。Okitsu小组[15]在合成(+)-2的过程中首先运用了该策略。首先酯(20)在三甲基铝酰胺的作用下很容易转化为Weinred酰胺(21),后经脱除羟基保护基,在DPPA作用下经Mitsunobu反应5-位羟基被叠氮基取代、叠氮基还原成胺基及Cbz保护氨基等反应合成(S)-5-胺基-N-苄氧羰基-2-羟基戊酸Weinreb酰胺(22),且构型保持。22在LiAlH4作用下还原胺化闭环,后经双乙酰化得哌啶中间体(24),收率24.8%。24与喹唑啉酮中间体缩合,水解得产物(+)-2(Scheme 3),收率4.3%。
该合成方法收率较低,工业化意义不大。
2.2 亲核取代法
1999 年,Kobayashi等[16]首次采用不对称合成法合成了2。该线路以D-谷氨酸(25)为原料经多步反应制得5-羟甲基-2-四氢呋喃酮(26);26经羟基保护、还原开环,TBS和苄基保护羟基及Swern氧化制得醛(28);28与甲基乙烯基甲醚和邻氨基苯甲醚在镱盐催化下经Mannich反应、亲核取代闭环及脱保护合成哌啶中间体(30),收率31%。30再经后续几步反应合成(+)-2,收率10.8%(Scheme 4)。
该合成方法的提出具有重要意义,不仅进一步证明了Barriger提出的2的相对构型的理论,同时确定了这类生物碱的绝对构型,开启了不对称合成2及其类似物的先河。
2009 年,Ashoorzadeh等[17]创新了一种(+)-常山碱合成线路。该线路以手性酸内酯(31)为原料在酸性条件下酯化开环,紧接苄基保护制得化合物33;33依次经LiAlH4还原及二苯磺酰化反应制得化合物35;35与羟胺盐酸盐经双亲核取代闭环反应制得重要中间体(S)-3-苄氧基-3,4,5,6-四氢吡啶-N-氧化物(37),收率 28.7%;随后与38经1,3偶极环加成,开环,氧化及脱保护等反应合成(+)-2,收率8.6%(Scheme 5)。
该线路证明了氮氧化物37可以作为哌啶生物碱合成过程中的重要手性-非外消旋合成砌块,具有1,3-偶极环加成立体选择性好、同末端烯进行环化反应收率高等优点,可有效制备反式-3-二取代-3-羟基哌啶和吲哚里西啶。用中间体37合成2及其衍生物将会使线路简短。
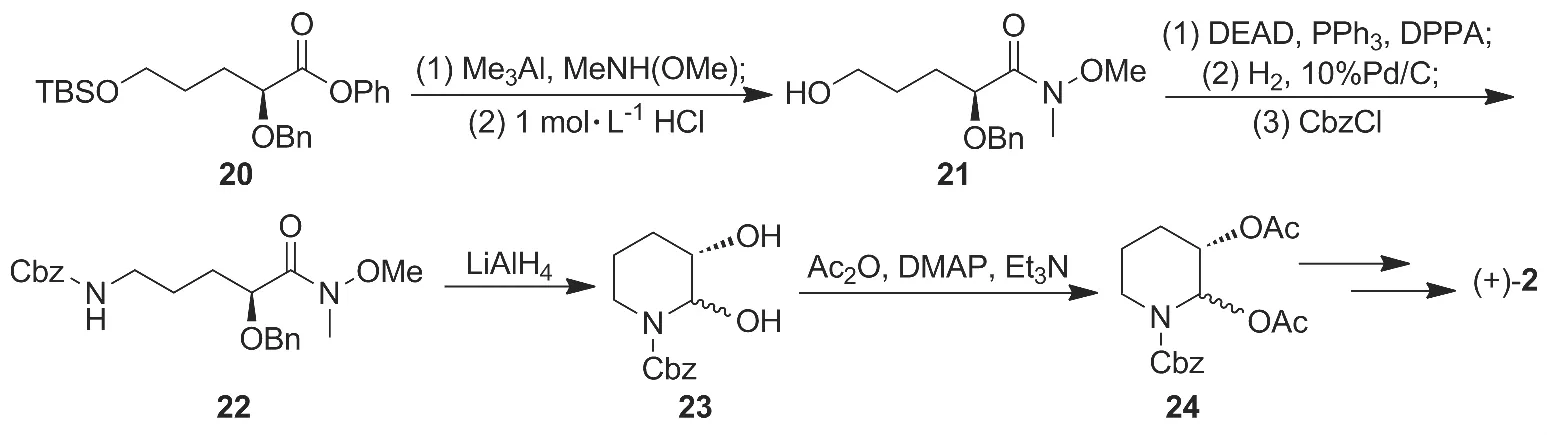
Scheme 3

Scheme 4
2009 年 Liu 等[18]以 L-谷氨酸-5-苄酯(39)为原料在酸性亚硝酸钠水溶液条件下脱除氨基后与甲醇反应制得酯(40);40经TBS保护羟基及5-位酯水解制得化合物41;41依次经还原和磺酰化后与叠氮钠反应制得42,后经Pd/C催化氢化还原和亲核取代环化形成酰胺(43);43经仲氮保护、硼氢化-氧化及酯化反应制得N,O-缩醛化合物(44);44在三氟化硼乙醚催化下经亲核取代反应制得高顺式非对映选择性化合物(45,顺/反=96/4),收率78%;45经溴代制得3的中间体46,后经缩合水解合成(+)-3(Scheme 6),收率 32.4%。
该线路收率高,每步工艺达到最优化状态,但整体线路过长,操作复杂,对设备要求高,大大提高了生产成本。
同年,Emmanuvel 等[19]以 1,5-己二烯-3-醇(47)为原料经Johnson-Claisen重排制得(E)-1,4-二烯酯(48)。后以(DHQ)2-PHAL为手性配体在四氧化锇催化下经不对称二羟化反应和酯交换制得化合物49;49的醇羟基经磺酰化制得磺酸酯,后经叠氮基取代制得化合物50;50经三苯基膦还原和取代反应制得内酰胺51;51经羟基氨基保护和溴代反应制得2的中间体54,收率34.9%;54经与喹唑啉酮缩合、氧化及脱保护等反应合成(+)-2(Scheme 7),总收率约15%。
该合成线路巧妙运用了Johnson-Claisen重排,不对称二羟化和亲核取代扩环反应进行哌啶环中间体的合成,线路短收率高,具有一定的工业生产意义,但催化剂四氧化锇毒性大且难以处理。

Scheme 5
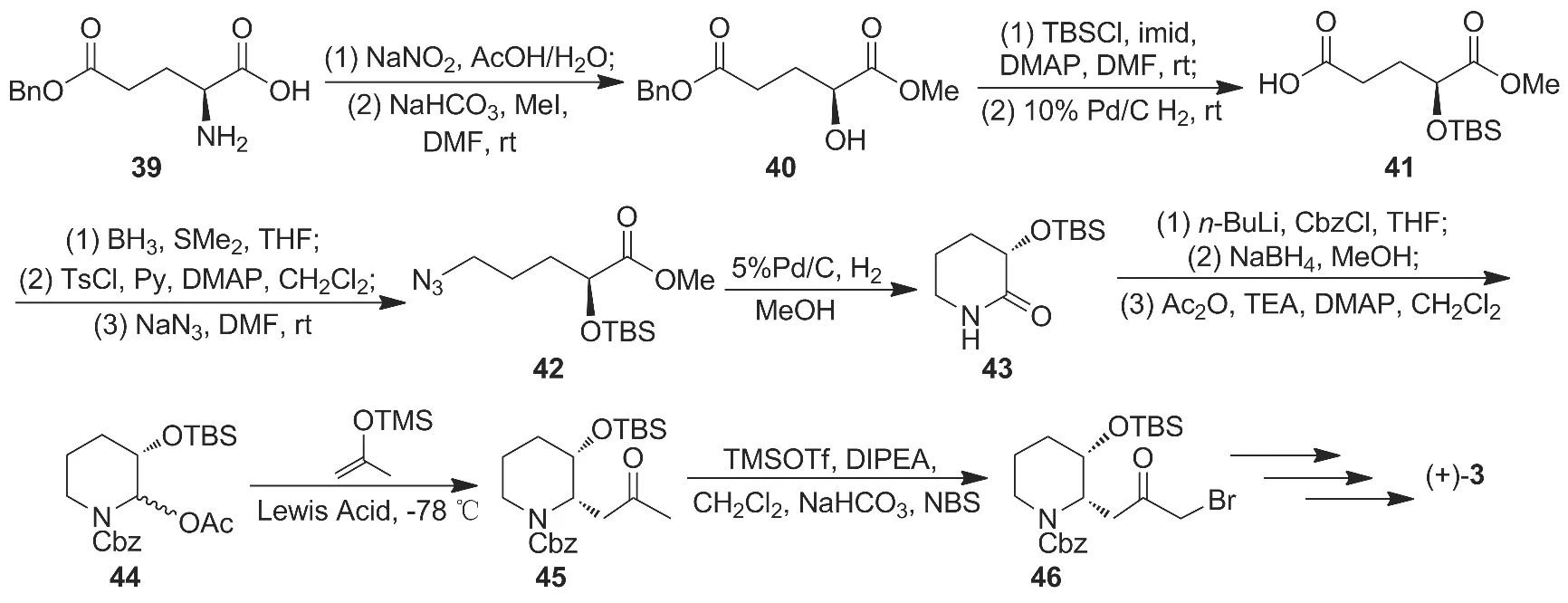
Scheme 6

Scheme 7
2013 年 Pansare 研究小组[20]以 γ-巴豆酰内酯(55)为原料,运用相同的闭环方式合成出了(+)-2和(+)-3。该线路以55为原料,以氨基芳酰胺(63)为催化剂经不对称插烯型羟醛反应制得化合物(56);56经催化氢化和DMP氧化制得57;57经K-Selectride还原制得58;58经磺酰化和叠氮化使得反应构型反转,后经催化氢化和碳酸钾重排制得化合物60;60经氢化铝锂(LAH)还原、仲氮保护、羟基保护及缩酮水解合成哌啶中间体(62),收率19.5%;62再经几步反应后最终合成(+)-2和(+)-3(Scheme 8),收率6.8%。
2.3 共轭加成法
Sukemoto等[21]通过共轭加成的方法获得了哌啶环骨架。以四氢哌啶为原料经仲氮保护和不对称双羟化制得化合物65;65经重排和Wittig反应制得开环化合物66;66在BF3·OEt2催化下经共轭加成制得trans-选择性单一异构体哌啶环中间体67。该哌啶中间体的合成巧妙运用了Wittig反应接上了链接部分碳链,通过共轭加成闭环得到哌啶中间体,收率33%;67经缩合水解合成2(Scheme 9),收率17.3%。
该合成路线是目前合成2的最短线路,收率高,具有一定的工业指导意义。
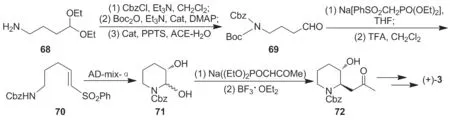
2010 年 McLaughlin[22]以同 Sukemoto 小组类似的成环方法,获得一条合成(+)-3的线路。该线路以缩醛化合物(68)为原料,经Cbz和Boc保护胺基,PPTS催化缩醛转化为醛(69);69经Horner-Wadsworth-Emmons反应和脱Boc保护制得α-丁烯砜(70);70经AD-mix-催化不对称双羟化反应制得71,ee 86%;后经HWE反应和分子内立体选择性共轭加成制得3-取代哌啶中间体72,收率41.3%;72经羟基保护、溴代、与喹唑啉酮中间体缩合及水解等反应合成(+)-3(Scheme 10),收率1.5%。
该线路两次灵活运用了HWE反应增长碳链,且AD-mix-催化的双羟化对映体选择高,但整体收率较低。

Scheme 8

Scheme 9
Scheme 10
Scheme 11
Scheme 12
3 关环复分解法
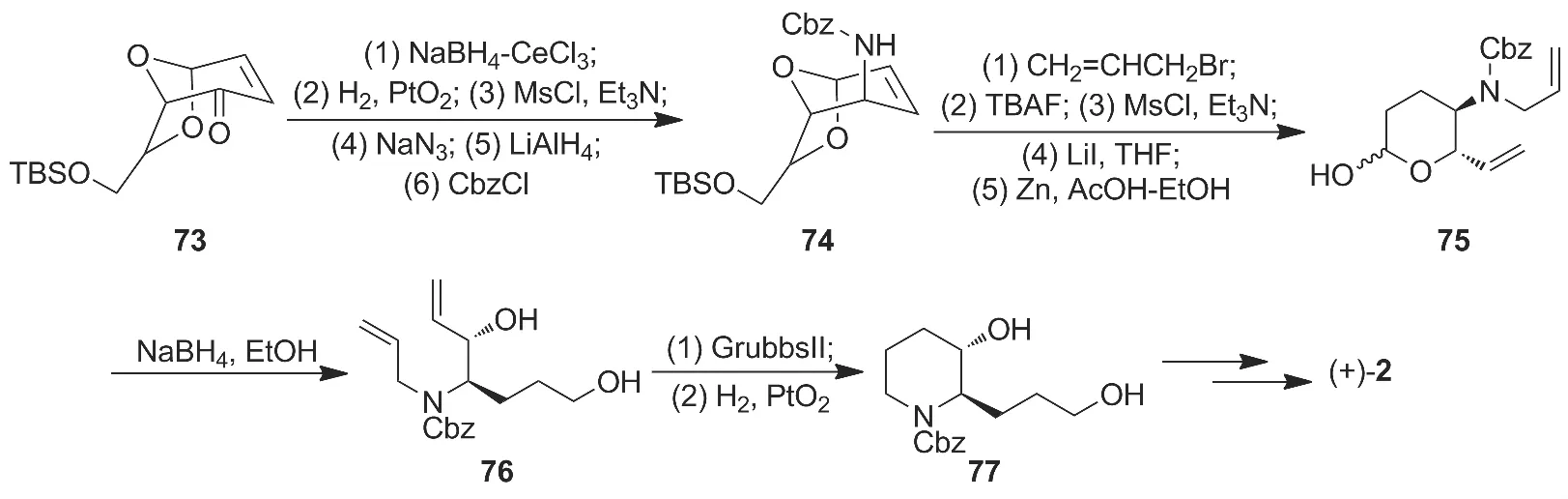
RCM反应是一种重要的闭环反应,Ogasawara研究小组[23]于2000年运用此策略合成了3的关键哌啶中间体。即以糠醛制备手性砌块(73),然后通过凸面-选择性1,2-还原制得内-烯丙基醇,随后羟基被叠氮基取代,叠氮基被还原后,经Cbz保护制得74;74经N-烯丙基化反应、去TBS保护、甲磺酰化、碘代和消除四步反应制得缩醛75;75与硼氢化钠反应制得二羟基二烯烃化合物(76);76在卡宾催化下经RCM反应和氢化还原制得哌啶环中间体77,收率36.9%;77再经多步反应最终合成(+)-2(Scheme 11),收率7.6%。
该合成路线共24步,线路冗长,没有生产上的实际意义。
4 扩环反应法
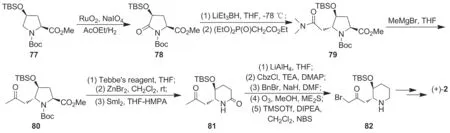
以吡咯烷衍生物为原料经区域选择性开环获得哌啶环体系是制备哌啶环衍生物的重要方法。该方法的优点在于多种重要的纯脯氨酸对映体均有市售。Katoh等[24]以反式-4-羟基-L-脯氨酸为原料全合成得到了(+)-2。首先,氨基、羟基和羧基分别被保护起来制得化合物77;77中吡咯烷环经钌氧化剂氧化和LiEt3BH局部还原后在NaH存在下经HWE反应制得具有手性活性的N-甲氧基-N-甲基甲酰胺(79);79经格氏加成和Tebbe试剂亚甲基化,Boc保护基、分子内还原耦合反应制得内酰胺81,后经LiAlH4还原,Cbz-保护氨基和Bn-保护羟基、臭氧断键及NBS溴代合成哌啶中间体82,收率20.1%;82经缩合和水解反应合成得(+)-2(Scheme 12),收率 18.83%。
该合成线路历经18步,收率较高,但步骤多,所用多种试剂昂贵,且多步反应需无水无氧操作,超低温反应,条件较苛刻,设备要求较高。
5 总结与展望
近年来随着生物医学技术的发展,人们逐渐认识到常山碱类化合物的生物活性,特别是几种在治疗慢性移植物抗宿主病、硬皮病、卡波稀释肉瘤、浅表性膀胱移行细胞癌及假肥大型肌营养不良症(DMD)上具有潜在活性的类似物,已成功进入临床试验阶段,这预示着科研和医疗对常山碱类化合物的需求将大幅增加。随着人们对常山碱及其类似物的合成探究的不断深入,多种3-羟基哌啶骨架中间体构建策略运用其中,将有效解决哌啶中间体合成难题,目前已开发出了多种具有工业生产价值的新生产工艺,这将能够有效满足未来对常山碱类化合物的需求。
[1]郭宗儒.天然产物的结构改造[J].药学学报,2012,47(2):144 -157.
[2]向甲甲,刘晓华,徐刚.苯二氮卓类药物检测方法[J].宁波化工,2014,1:6 -9.
[3]贺能琴,严胜骄,林军.喹唑啉类化合物的合成及活性研究进展[J].化学通报,2010,4:314 -323.
[4]McLaughlin N P,Evans P,Pines M.The chemistry and biology of febrifujine and halofugunone[J].Bioorganic & Medicinal,2014,22:1993 -2004.
[5] 叶晶晶,殷浩,孙波,等.桑树中1-脱氧野尻霉素含量变化规律研究蚕叶科学[J].2009,35(4):722 -727.
[6]Heimgartner G,Raatz D,Reiser O.Stereoselective synthesis of swainsonines from pyridines[J].Tetrahedron,2005,61:643 -655.
[7]Ruan S T,Luo J M,Du Y,et al.Asymmetric vinylogous Mannich reaction:A versatile approach functionalized heterocycles[J].Org Lett,2011,13(18):4938-4941.
[8]Jouanno L A,Tognetti V,Joubert L,et al.Thermally controlled decarboxylative[4+2]cycloaddition between alkoxyoxazoles and acrilic acid:Expedient access to 3-hydroxypyridines[J].Org Lett,2013,15(10):2530-2533.
[9]Tone L H,Guan Q N,Christopher Y C,et al.Halofuginone suppresses T cell proliferation by blocking proline uptake and inducing cell apoptosis[J].International Immunopharmacology,2013,16:414 -423.
[10]Broekaert N,Daeseleire E,Delezie E,et al.Can the use of coccidiostats in poultry breeding lead to residues in vegetables?An experimental study[J].Journal of Food Chemistry,2012,60:12411 -12418.
[11]李燕,刘明川,金林红,等.常山化学成分及生物活性研究进展[J].广州化工,2011,39(9):7 -9.
[12]Kikuchi H,Horoiwa S,Kasahara R,et al.Synthesis of Febrifugine derivatives and development of an effective and safe tetrahydroquinazoline-type antimalarial[J].European Journal of Medical Chemistry,2014,76:10-19.
[13]顾艳艳,刘世旭,江涛,等.常山酮的合成方法研究[J].中国海洋大学学报,2011,41(9):67 -70.
[14] 吴世林,张贵东,梁庭枝,等.一种2-丙酮基-3-甲氧基哌啶的制备方法[P].中国103 664 741 A,2014.
[15]Okitsu O,Suzuki R,Kobayashi S.Efficient synthesis of piperidine derivatives.Development of metal triflate-catalyzed diastereoselective nucleophilic substitution reactions of 2-metheoxy-and 2-acyloxypiperidines[J].J Org Chem,2001,66:809 -823.
[16]Kobayashi S,Ueno M,Suzuki R,et al.Catalytic asymmetric synthesis of antimalarial alkaloids Febrifugine and isofebrifugine and their biological activity[J].J Org Chem,1999,64:6833 -6841.
[17]Ashoorzadeh A,Archibald,Caprio V.Synthetic evaluation of an enantiopure tetrahydropyridine N-oxide.Synthesis of(+)-Febrifugine[J].Tetrahedron,2009,65:4671 -4680.
[18] Liu R C,Huang W,Ma J Y,et al.BF3·Et2O catalyzed diastereoselective nucleophilic reactions of 3-silyloxypiperidine N,O-acetal with silyl enol ether and application to the asymmetric synthesis of(+)-Febrifugine[J].Tetrahedron Letters,2009,50:4046 - 4049.
[19]Emmanuvel L,Kamble D A,Sudalai A.A concise enantioselective synthesis of(+)-Febrifugine[J].Tetrahedron:Asymmetry,2009,20:84 -89.
[20]Pansare S V,Paul E K.Synthesis of(+)-febrifugine and a formal synthesis of(+)-halofuginone employing an organocatalytic direct vinylogous Aldol Reaction[J].Synthesis,2013,45:1863 -1869.
[21]Sukemoto S,Oshige M,Sato M,et al.Concise asymmetric synthesis of(+)-Febrifugine utilizing trans-selective intramolecular conjugate addition[J].Synthesis,2008,19:3081 -3087.
[22]McLaughlin N P,Evans P.Dihydroxylation of vinyl sulfones:Stereoselective synthesis of(+)-and(- )-Febrifugine and halofuginone[J].J Org Chem,2010,75:518 -521.
[23]Taniguchi T,Ogasawara K.A diastereocontrolled synthesis of(+)-Febrifugine:A ptent antimalarial piperidine alkaloid[J].Org Lett,2000,2(20):3193 - 3195.
[24]Katoh M,Matsune R,Nagase H,et al.Stereocontrolled synthesis of a potent antimarial alkaloid,(+)-febrifugine[J].Tetrahedron Letters,2004,45:6221-6223.
